Key points
- The PI3K/AKT/mTOR complex plays a significant role in the regulation of cellular growth by controlling different processes in protein synthesis and angiogenesis. Dysregulation of this pathway is commonly found in kidney cancer, breast cancer and neuroendocrine tumours.
- Currently, there are two mTOR inhibitors commercially available in the UK and the United States: temsirolimus and everolimus. Temsirolimus is approved by the European Medicines Agency (EMA) for advanced renal cell carcinoma (RCC) and mantle cell lymphoma; in the United States, temsirolimus is only indicated for RCC. Everolimus is EMA-approved and approved by the US Food and Drug Administration for advanced RCC, breast cancer and pancreatic neuroendocrine tumours.
- Common and serious class side effects of mTOR inhibitors include non-infectious pneumonitis, metabolic disorders and mucosal toxicity.
- Despite progression-free and overall survival benefit, response to mTOR inhibitors is not durable and patients ultimately progress because of various mechanisms of resistance.
- Novel agents, designed to inhibit multiple targets within the PI3K/AKT/mTOR pathway, are under investigation with the intent to overcome emerging resistance. Some have already begun to show promising results in cancer cell lines and xenograft models.
Introduction
The PI3K/AKT/mTOR signalling pathway and its role in cancer
Source: Wikimedia Commons
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase of the PI3K/AKT signalling pathway. Stimulation of PI3K, via nutrients, hormones and various growth factors, leads to activation of downstream pathways that involve AKT and mTOR, resulting in cell proliferation, growth, protein synthesis and angiogenesis[1]
. mTOR regulates protein synthesis and promotes translation of mRNAs. mTOR exists as two multiprotein complexes: mTOR complexes 1 and 2 (mTORC1 and mTORC2). Growth factors and nutrients stimulate mTORC1 activity by triggering receptor tyrosine kinase dimerisation and downstream autophosphorylation[2]
. mTORC1 acts as a central regulator of protein synthesis. It stimulates initiation of mRNA translation by phosphorylating vital components of the translation mechanism: the eukaryotic initiation faction 4E (eIF4E)-binding protein (4E-BPs) and the ribosomal S6 kinases (S6Ks) 1 and 2. The PI3K/AKT/mTOR pathway plays a critical role in cancer cell metabolism[1]
. Research has demonstrated that the PI3K pathway regulates cellular proliferation, growth, apoptosis and cytoskeletal rearrangement. Phosphatase and tensin homolog (PTEN) is a tumor suppressor gene that functions as a negative regulator of the PI3K/ATK/mTOR signalling pathway[3]
. Loss of PTEN function is commonly implicated in various cancers, which results in uninhibited activation of the PI3K pathway leading to uncontrolled cell proliferation. Deregulation of this PI3K/AKT/mTOR pathway has been demonstrated in several solid malignancies, including kidney, breast and endometrial. Therapeutic targets of this pathway are available and in development as cancer therapeutics.
mTOR Inhibitors as anticancer therapies
mTOR inhibition in several solid malignancies, such as renal cell carcinoma, neuroendocrine carcinoma, breast cancer, glioblastoma multiforme and endometrial cancer, has shown promising progression-free and overall survival in clinical studies[4],[5],[6],[7],[8]
. There is also efficacy data in haematological malignancies, including Hodgkin’s lymphoma, mantle cell lymphoma and Waldenström macroglobulinaemia, however that is beyond the scope of this review[9],[10],[11],[12],[13]
. Currently, two mTOR inhibitors are approved for use in Europe and the United States and are commercially available: temsirolimus and everolimus.
Summary of clinical trials
Renal cell carcinoma
Historically, renal cell carcinoma (RCC) has been relatively resistant to traditional chemotherapy. With the advent of novel targeted therapy, there have been modest improvements in progression free survival and overall survival for this otherwise chemotherapy-resistant disease. Both temsirolimus and everolimus have EMA-approved and FDA-approved indications for the treatment of renal cell carcinoma.
The pivotal phase III study of temsirolimus enrolled a total of 626 patients, who were randomised to three different treatment arms: interferon, temsirolimus, or a combination of interferon and temsirolimus[5]
. Compared with interferon alone, treatment with temsirolimus was associated with a hazard ratio [HR] for death of 0.73 (95% confidence interval [CI] 0.58–0.92; P =0.008). Treatment with a combination of temsirolimus and interferon was similar when compared with treatment with interferon alone (HR=0.96; 95% CI 0.76–1.2; P =0.7). Median survival was 7.3 months in the interferon group, 10.9 months in the temsirolimus group, and 8.4 months in the combination group. The benefit of temsirolimus was most prominent in patients with at least three poor prognostic factors. These predictors included elevated serum lactate dehydrogenase, anaemia, hypercalcaemia, poor performance status, multiple sites of metastases, and less than one year of time from initial diagnosis to randomisation[14],[15]
. Based on the efficacy and safety of temsirolimus established in this trial, temsirolimus is currently recommended as first-line therapy for poor-risk patients with unresectable or relapsed RCC[5],[16],[17],[18],[19]
.
The phase III trial RECORD-1 established the role of everolimus in patients with advanced RCC who had progressed after treatment with sunitinib, sorafenib or both[20]
. Patients were randomly assigned in a 2:1 ratio to receive everolimus 10mg once daily or placebo, both in conjunction with best supportive care. Progression-free survival (PFS) was the primary endpoint and patients were assessed for efficacy on an intention-to-treat analysis. PFS was prolonged in the everolimus group compared with the placebo-group (HR=0.3; 95% CI 0.22–0.4; P <0.0001), assessed by a blinded independent central review. Median PFS was four months in everolimus group and 1.9 months in the placebo group. The most common adverse effects, all grades, reported in the everolimus group (n=269) were stomatitis (40%), rash (25%), fatigue (20%), asthenia (18%), and diarrhoea (17%). Patients who received everolimus had higher rates of grade three or four stomatitis (3% vs 0%), infections (3% vs 0%), and non-infectious pneumonitis (3% vs 0%) than those in the placebo group. This study supports the use of everolimus as second-line treatment after progression with VEGF-targeted therapies in advanced RCC.
With promising improvement of PFS in the second-line setting, everolimus was compared with sunitinib as first-line therapy in a phase II non-interiority head-to-head trial, RECORD-3[21]
. Patients were randomly assigned to receive first-line everolimus followed by sunitinib, or first-line sunitinib followed by everolimus. If patients had documented disease progression, they were allowed to crossover to second-line sunitinib or everolimus. Everolimus was given as 10mg daily continually, and sunitinib was 50mg daily four weeks on, followed by two weeks off. Although everolimus did not demonstrate noninferiority compared with sunitinib, everolimus did not meet the study’s parameters for inferiority to sunitinib as first-line therapy. Median PFS was shorter for first-line everolimus (7.9 months) than for first-line sunitinib (10.7 months). Median PFS was 21.1 months for combined first-line everolimus and second-line sunitinib, and 25.8 months for combined first-line sunitinib and second-line everolimus (HR=1.3; 95% CI 0.9–1.7). Median overall survival was 22.4 months for first-line everolimus and second-line sunitinib, and 32 months for first-line sunitinib and second-line everolimus (HR=1.2; 95% CI 0.9–1.6). Patients with favourable and intermediate risk (based on the Memorial Sloan Kettering Cancer Center prognostic score)[14],[15]
and those with clear-cell and non-clear cell histology had longer median PFS when treated with first-line sunitinib than with first-line everolimus. Side effects were consistent with the known safety profiles of everolimus and sunitinib. The most common adverse events in the group that received everolimus as first-line therapy (n=238) were stomatitis (53%), fatigue (45%), diarrhoea (38%), cough (38%), and rash (37%). The study results support the current treatment algorithm of first-line sunitinib, followed by everolimus at disease progression in the United States. However, the National Institute for Health and Care Excellence (NICE) in the UK does not recommend everolimus as second-line therapy for patients because it did not provide enough benefit to patients to justify its high cost[22]
.
Although the combination of mTOR inhibitors and other targeted therapies have not demonstrated survival benefit and are associated with significant toxicities[18]
, the combination of temsirolimus and lenvatinib (an oral multitarget tyrosine kinase inhibitor) has demonstrated survival benefit and seems to be relatively well tolerated[23]
. In addition, two randomised phase III trials recently published results comparing everolimus with cabozantinib (METEOR)[24]
and with nivolumab (Checkmate 025)[25]
in advanced RCC. In the METEOR trial, median PFS was significant higher with cabozantinib than with everolimus among patients who progressed after VEGFR-targeted therapy (7.4 months vs 3.8 months, respectively). In the Checkmate 025 trial, nivolumab was found to be superior to everolimus in previously treated patients with advanced clear-cell renal carcinoma. Median overall survival (OS) was 25 months with nivolumab and 19.6 months with everolimus (HR for death=0.73; 98.5% CI 0.57–0.93; P =0.002). Given these promising results in the METEOR and Checkmate 025 trials, the preferred treatment options for patients may begin to shift away from mTOR inhibitors towards treatment with novel multitargeted kinase agents, and/or with immunotherapy.
Breast cancer
The PI3K/AKT pathway plays an important role in breast cancer. Upregulation of this pathway by human epidermal growth factor receptors (HER), insulin-like growth factors and oestrogen receptors increases the activity of mTOR. This can also lead to development of resistance to endocrine therapy, cytotoxic chemotherapy and trastuzumab[26],[27],[28]
. The mechanism of resistance is believed to occur via complex molecular cross-talk between the oestrogen receptor and growth factor receptor signalling pathways. Therefore, the addition of mTOR inhibitors to existing breast cancer therapies may help overcome acquired resistance and restore tumour sensitivity. Everolimus is EMA-approved and FDA-approved for advanced hormone receptor (HR)-positive, HER-2 negative breast cancer in combination with exemestane after treatment failure with endocrine therapy. In breast cancer, trastuzumab resistance is linked with loss of PTEN, resulting in unregulated activation of the PI3K/AKT/mTOR signalling pathway[29],[30],[31]
. Combination therapy with everolimus and trastuzumab in patients with HER2 positive, metastatic breast cancer who progressed on trastuzumab-based therapy is also being investigated in clinical trials[32]
.
The pivotal, phase III BOLERO-2 trial enrolled 724 postmenopausal women with HR-positive, HER-2 negative, advanced breast cancer who were refractory to letrozole or anastrozole[33]
. Patients were randomised to either everolimus plus exemestane, or placebo plus exemestane. The primary endpoint was PFS. Patients received everolimus 10mg daily, or placebo, with exemestane 25mg daily. Median PFS, assessed by local investigators, was 6.9 months and 2.8 months (HR=0.43; 95% CI 0.35–0.54; P <0.001) for everolimus plus exemestane and placebo plus exemestane, respectively. Central assessment demonstrated consistent results with a median PFS of 10.6 months for everolimus compared with 4.1 months with placebo (HR=0.36; 95% CI 0.27–0.47; P <0.001). This combination demonstrated significant improvement in PFS for patients with HR-positive, HER-2 negative, advanced breast cancer who had failed treatment with aromatase inhibitors. The two most common reasons for treatment discontinuation were progression rate (37% in the combined and 66% in the placebo group) and adverse events (19% vs 4%). Patients who received everolimus had higher rates of grade three and four stomatitis (8% vs 1%), anaemia (6% vs <1%), dyspnoea (4% vs 1%), hyperglycaemia (4% vs <1%), fatigue (4% vs 1%), elevated alanine aminotransferase (3% vs 2%), elevated aspartate aminotransferase (3% vs 1%), and pneumonia (3% vs 0%).
Updated results of the BOLERO-2 were consistent with previously reported results[34]
. Median PFS, assessed by local investigators, was 7.8 months for everolimus plus exemestane group compared with 3.2 months in the placebo plus exemestane group (HR=0.45; 95% CI 0.38–0.54; P <0.0001). Median PFS, assessed by central investigators, was 11 months for everolimus plus exemestane and 4.1 months for placebo plus exemestane (HR=0.38; 95% CI 0.31–0.48; P =0.0001). The main reason for discontinuation of treatment was disease progression (61.9% and 88.7% in the combined and placebo group, respectively). The adverse event profile in this analysis is consistent with the pivotal phase III BOLERO-2 trial. Patients who received everolimus had higher rates of grade three and four stomatitis (8% vs 0%), fatigue (4% vs 1%), dyspnoea (5% vs <1%), anaemia (7% vs <1%), and hyperglycaemia (5% vs <1%). The BOLERO-2 updates confirmed that everolimus plus exemestane provide a significant benefit in PFS in advanced breast cancer HR-positive, HER-2 negative patients. Therefore, the National Comprehensive Cancer Network (NCCN) states that this combination can be considered for patients who meet the eligibility criteria for BOLERO-2[35]
. However, NICE and the Scottish Medicines Consortium (SMC) do not recommend its use given the high cost-to-benefit ratio[36],[37]
.
A sub-analysis was done on the safety and efficacy of the everolimus and exemestane combination in elderly patients in the BOLERO-2 trial[34]
. A total of 275 patients aged 65 years and 164 patients aged 70 years participated in the BOLERO-2 trial. Median PFS was 8.11 months in the everolimus group and 4.01 months in the placebo group in patients aged <70 years (HR=0.44; 95% CI 0.36–0.54). In patients aged 70 years, median PFS were 6.77 months (HR=0.45; 95% CI 0.30–0.68) and 1.51 months in the everolimus group and the placebo group, respectively. At the time of this analysis, the median PFS for all ages was 7.8 months in the everolimus group and 3.2 months in the placebo group (HR=0.45; 95% CI 0.38–0.54; P <0.0001). In elderly patients aged ≥ 70 years, the addition of everolimus reduced PFS events by 55% (HR=0.45; 95% CI 0.30–0.68) compared with the placebo group. The sub-analysis demonstrated the efficacy and safety data in the elderly patients were comparable to all patients of all ages. In elderly patients aged ≥70 years, the most common adverse events, all grades, reported in the everolimus group (n=164) were stomatitis (49%), fatigue (38%), decreased appetite (36%), and diarrhoea (36%). In the subset analysis of elderly patients aged ≥75 years, the most common grade three and four adverse events in the everolimus group were fatigue (14%), stomatitis (10%), dyspnoea (10%), and anaemia (10%). Although there was a higher rate of adverse events in the elderly group, dose modifications were similar in elderly and younger patients reported in the everolimus group.
Pancreatic neuroendocrine tumours
Insulin-like growth factor 1 (IGF-1) stimulates the mTOR pathway, resulting in tumour proliferation[38],[39],[40]
. Pancreatic neuroendocrine tumours (PNET) are known to overexpress IGF-1. This makes mTOR inhibitors an appealing targeted treatment option for PNET. The placebo-controlled RADIANT-3 trial demonstrated significantly longer PFS in patients with pancreatic neuroendocrine tumours. Median PFS was 11 months with everolimus, compared with 4.6 months with placebo (HR=0.35; 95% CI 0.27–0.45; P <0.001). Around 5% of patients in the everolimus group had an objective tumour response (all partial responses), compared with 2% in the placebo group. Stable disease was evident in 73% of the patients in the everolimus group, compared with 51% in the placebo group. A total of 64% of the patients receiving everolimus experienced some degree of tumour shrinkage, compared with 21% in the placebo group. Of the patients assigned to the placebo arm, 73% eventually crossed over to open-label everolimus. Median OS was not reached at the time of study analysis[8]
.
The results of the phase III study, RADIANT-2, further confirmed PFS benefit with everolimus in the treatment of advanced neuroendocrine tumours[40]
. The study randomised 429 patients with neuroendocrine tumors to a combination of everolimus and long-acting octreotide or to octreotide alone. The combination of everolimus and octreotide was associated with an increase in median PFS of 12 months versus 8.6 months (HR=0.78; 95% CI 0.62–0.98; P <0.018), a reduction in relative risk of progression of 22%. Overall, 75% of the patients in the everolimus plus octreotide LAR group and 45% in the placebo plus octreotide LAR group experienced tumour shrinkage. Compared with 81% in the octreotide LAR alone group, stable disease was evident in 84% of the patients in the everolimus plus octreotide LAR group. Median OS was not reached at the time of study analysis since 123 of 213 patients (58%) in the octreotide alone arm crossed over to receive everolimus. Given the success of everolimus in PNET, RADIANT-4 is an ongoing phase III trial evaluating everolimus versus best supportive care in the treatment of patients with advanced nonfunctional neuroendocrine tumours of the gastrointestinal tract or lung (Clinicaltrials.gov, NCT01524783)[41]
.
| Table 1: Pharmacological properties of temsirolimus and everolimus | ||
|---|---|---|
| Temsirolimus | Everolimus | |
| Commercial names | Torisel®, CCI-779 | Afinitor, RAD001 |
| Mechanism of action | Inhibits the mammalian target of rapamycin (mTOR) kinase, which leads to the prevention of protein synthesis, cell proliferation and angiogenesis | |
| FDA- & EMA-approved indications | Renal cell carcinoma (FDA), as first-line treatment of adults with advanced renal cell carcinoma, with at least six prognostic factors (EMA) | Renal cell carcinoma, b reast cancer, p ancreatic neuroendocrine tumour, s ubependymal giant cell astrocytoma |
| Off-labelled indications | Glioblastoma multiforme, m elanoma, mantle cell lymphoma | Waldenström macroglobulinaemia, c arcinoid tumours |
| Usual dosing | 25mg IV infused over 30–60 minutes once weekly. Premedicate with diphenhydramine 25–50mg IV (or similar antihistamine) 30 minutes prior to each dose | 10mg PO daily |
| Absorption | Tmax (temsirolimus) 0.5 hours, Tmax (sirolimus) 1–2.5 hours | Tmax 1–2 hours, h igh-fat meal reduced AUC and Cmax |
| Distribution | Vd at steady state: 172 L, Vd (sirolimus) 622–1732 L depending on dose | Vd = 102–342 L, p rotein binding = 74% |
| Metabolism | Hepatic; extensively metabolised via CYP3A4 to its active metabolite: sirolimus | Hepatic: CPY3A4 and P-gP |
| Elimination | Faecal 78%, r enal 4.6% | Faecal 80%, r enal 5% |
| Terminal half-life | Temsirolimus 17.3 hours, s irolimus 54.6 hours | 30 hours, m oderate hepatic impairment: 79 hours |
| Renal dose adjustment | No dosage adjustment necessary. Both temsirolimus and sirolimus are not dialysable | No dosage adjustments recommended |
| Hepatic dose adjustment | Mild impairment: 15mg IV once a week. Contraindicated in bilirubin >1.5 ULN | Mild (Child-Pugh class A): reduce dose to 7.5mg daily; if not tolerated reduce to 5mg daily. Moderate (Child-Pugh class B): reduce dose to 5mg daily; if not tolerated reduce to 2.5mg daily. Severe (Child-Pugh class C): use only if benefit outweighs risk; do not exceed dose of 2.5mg daily |
| Drug interactions a | Avoid if possible or consider dose reduction of temsirolimus to 12.5mg/wk with concurrent strong inhibitors of CYP3A4. Avoid if possible or consider dose increase of temsirolimus from 25mg/wk to 50mg/wk with concurrent strong inducers of CYP3A4 | Concomitant use of moderate CYP3A4 and/or P-glycoprotein inhibitor; reduce dose to 2.5mg daily, increasing to 5mg daily if tolerated. Concomitant use of strong CYP3A4 and/or P-glycoprotein inducer; if use cannot be avoided, consider doubling the daily everolimus dose in 5mg increments or less |
a Strong CYP3A4 inhibitors include ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, voriconazole, grapefruit juice; CYP3A4 inducers include rifampin, phenytoin, carbamazepine, rifabutin, rifampacin, phenobarbital[42],[46] | ||
Temsirolimus and everolimus: pharmacology
Approved indications and recommended dosing
For pharmacological properties, see ‘Table 1: Pharmacological properties of temsirolimus and everolimus’. Temsirolimus (Torisel) was approved by the FDA and the EMA in 2007 for the treatment of advanced RCC[42]
. Of note, the EMA indication for temsirolimus is as first-line treatment of adult patients with advanced RCC who have at least three of the six prognostic risk factors; the FDA-indication does not have this particular distinction[43]
. The dose for advanced RCC is 25mg once weekly infused intravenously over 30–60 minutes. Patients should receive prophylactic intravenous diphenhydramine 25–50mg (or a similar antihistamine) approximately 30 minutes prior to each dose of temsirolimus because of the risk of infusion-related reactions. Treatment is continued until the disease progresses or until there is unacceptable toxicity. Dose reduction is required in patients with mild hepatic impairment. Temsirolimus is typically scheduled to be given once weekly, or daily for five days every two weeks. Dose finding trials indicate that antitumour activity can be achieved with intermittent dosing while minimising its immunosuppressive effects[44],[45]
.
Everolimus (Afinitor) was approved for use by the EMA and the FDA in 2009. It is currently indicated in breast cancer, PNET and advanced RCC in Europe and in the United States[46]
. The recommended dose is 10mg once daily. Everolimus is also indicated for the treatment of paediatric and adult patients with tuberous sclerosis complex (TSC) who have subependymal giant cell astrocytoma (SEGA). The recommended dose for this indication is 4.5mg/m2 once daily. Doses should be adjusted to attain trough concentrations of 5–15ng/mL in patients with TSC and SEGA. Therapeutic drug monitoring is not necessary when everolimus is used to treat solid malignancies. Everolimus is available as 2.5mg, 5mg, 7.5mg and 10mg tablets. It is also available as tablets for oral suspension (Afinitor Disperz) in 2mg, 3mg and 5mg strengths for paediatric patients.
Mechanism of action
mTOR inhibitors function by forming a complex with the intracellular protein FKBP-12, which then fuses with mTORC1 (see figure 1)[47],[48]
. This complex inhibits mTOR by preventing it from interacting with the raptor binding protein and interrupting further cell signalling downstream. This immediately stops downstream phosphorylation of 4EBP1 (a translation initiation factor) and S6K1 (a ribosomal protein); inactivation of these proteins leads to reduced protein translation and synthesis related to transitioning cells from G1 to S phase in the cell cycle. Progression in the cell cycle is also affected by mTOR inhibitors decreasing the translation of cell cycle regulators like cyclin D1, which result in cell stasis at the G1 phase. Another proposed mechanism of mTOR inhibitors is the ability to regulate angiogenesis by monitoring the translation of hypoxia-inducible factors (HIF). Inhibition of HIF results in decreased VEGF, platelet-derived growth factors (PDGF), HER and other growth factors related to the promotion of angiogenesis[49]
. It is a combination of these molecular mechanisms of mTOR inhibitors that ultimately prohibit cell proliferation, growth and angiogenesis in tumour cells.
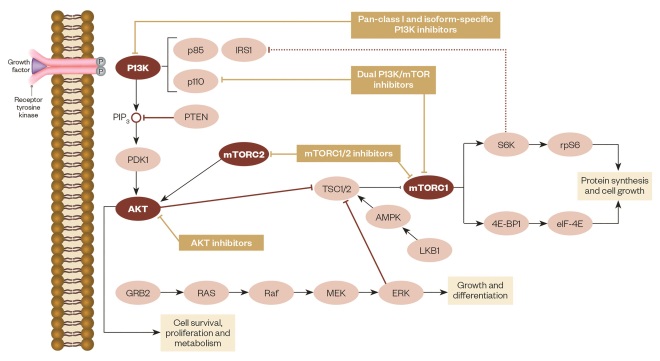
P13K/AKT/mTOR pathway
Source: Adapted from Martelli AM, Chiarini F, Evangelisti C et al. Oncotarget 2012;3(4):371–394.
Pharmacokinetics: ADME of temsirolimus and everolimus
Temsirolimus is primarily metabolised via the CYP3A4 to its main active metabolite sirolimus[42],[50]
. However, it is not a prodrug because temsirolimus does have intrinsic mTOR inhibitory activity. The mean half-life of temsirolimus and sirolimus is 17.3 hours and 54.6 hours, respectively. Temsirolimus is primarily eliminated through the faeces (80%), while 5% is excreted in the urine. Peak concentrations of everolimus are reached one to two hours after oral administration[46]
. Steady-state is achieved within two weeks following once-daily dosing. Although high-fat meals delay time to peak concentration (Tmax) and reduce systemic exposure, it is recommended that everolimus is taken consistently with or without food to reduce variability[46],[51],[52]
. Everolimus is a substrate of CYP3A4 and PgP. The mean elimination half-life of everolimus is approximately 30 hours. Around 80% of everolimus metabolites are eliminated through the faeces, and 5% is excreted in the urine.
Dosage adjustments and special patient populations
Renal impairment
Patients with RCC may require haemodialysis on account of disease. Lunardi et al.
[53],[54]
compared the pharmacokinetics of temsirolimus and its metabolite, sirolimus, among patients with RCC receiving haemodialysis to those not receiving haemodialysis. The study did not find a statistically nor a clinical significant difference of temsirolimus pharmacokinetics between patients on haemodialysis and those who were not. Therefore, temsirolimus does not require dosing adjustments for renal insufficiency or if a patient requires haemodialysis[42]
. Similarly to temsirolimus, everolimus is mainly eliminated through the faeces with minimal renal excretion; thus, dose adjustments in patients with renal insufficiency is also unnecessary[46]
.
Hepatic impairment
The dose escalation phase I study of temsirolimus[55]
found that patients with moderate and severe hepatic impairment (Child-Pugh class B and C, respectively) had increased rates of adverse reactions. Therefore, temsirolimus is contraindicated in patients with a bilirubin greater than 1.5 times the upper limit of normal (ULN). It is recommended that the dose of temsirolimus is reduced to 15mg/week in patients with mild hepatic impairment, defined as a bilirubin more than 1–1.5 times ULN, or if the AST is more than the ULN but bilirubin less than or equal to the ULN. European labelling states that for patients with RCC and severe hepatic impairment, the recommended dose for patients who have baseline platelets >100x 109/L is 10mg/week[43]
. The pharmacokinetics and safety of everolimus were evaluated in healthy volunteers with normal hepatic function and in patients with hepatic impairment[56]
. Compared with healthy subjects, everolimus concentrations were increased in patients with mild (Child-Pugh class A), moderate (Child-Pugh class B), and severe hepatic impairment (Child-Pugh class C). For those with mild or moderate hepatic impairment, a dose reduction to 7.5mg/day and 5mg/day, respectively, should be considered[46],[56]
. However, everolimus is not recommended in patients with severe hepatic impairment, unless benefits overweigh the risk; in this case, the recommended maximum daily dose of everolimus should not exceed 2.5mg/day.
Effects of age and gender
In phase I and pharmacokinetic studies of temsirolimus and everolimus, no apparent age-related or gender-related effects were found[50],[57]
. However, in the BOLERO-2 study in advanced breast cancer, adverse reactions leading to discontinuation were increased in patients aged 65 years and over, compared with patients aged under 65 years (15.9% vs 4.5%). The incidence of deaths of any cause within 28 days after treatment was also increased in patients aged 65 years or over than those aged under 65 years (6.8% vs 2.1%)[37],[58]
. Therefore, careful monitoring for side effects and appropriate dose adjustments of mTOR inhibitors may be warranted in older patients.
Effects of ethnicity
In a phase I study of temsirolimus in Japanese patients with advanced cancer, Japanese patients had on average higher systemic exposures than non-Japanese patients receiving the same dose[42],[59]
. No significant difference in clearance was detected for Asian patients, whereas black patients had an average clearance that was 20% higher than that of non-black patients[57]
. Despite these observations, the implications and significance of these differences in Japanese or in black patients have not been established[42]
. Therefore, there are no formal dosing recommendations or adjustments based on ethnicity.
Drug interactions
Temsirolimus
Although coadministration with ketoconazole, a potent CYP3A4 inhibitor, does not affect temsirolimus, coadministration does affect its major metabolite, sirolimus. Coadministration with ketoconazole resulted in a greater than 200% increase in the maximum concentration (Cmax) and the area under the curve (AUC) and a 69% reduction in its clearance — thus, a clinically significant drug interaction[60],[61]
. It is recommended that concomitant use of strong CYP3A4 inhibitors should be avoided[42]
. If coadministration of an inhibitor cannot be avoided, a dose reduction to 12.5mg/week should be considered. Coadministration with strong CYP3A4 inducers should also be avoided. Again, these recommended dose adjustments are based on pharmacokinetic studies, and their clinical implications on efficacy have not been elucidated. If the inhibitor or inducer is discontinued, the temsirolimus dose should be returned to the dose used prior to any inhibitor or inducer. Grapefruit and other foods known to inhibit CYP450 and PgP activity may increase exposure to temsirolimus and should be avoided during treatment.
Everolimus
It is recommended that use of strong CYP3A4/PgP inhibitors (e.g. ketoconazole, itraconazole, clarithromycin, atazanavir, nefazodone, saquinavir, telithromycin, ritonavir, indinavir, nelfinavir, voriconazole) is avoided. Use caution when coadministered with moderate CYP3A4/PgP inhibitors (e.g. amprenavir, fosamprenavir, aprepitant, erythromycin, fluconazole, verapamil, diltiazem)[42],[62],[63]
. If patients require a moderate CYP3A4/PgP inhibitor, a dose reduction to 2.5mg daily may be considered. The reduced dose can be increased to 5mg based on patient tolerance. The use of concomitant strong CPY3A4/PgP inducers should also be avoided. Providers can consider doubling the daily dose of everolimus using increments of 5mg or less. Of note, these dosage adjustment recommendations are based on predictive pharmacokinetics models, and not on clinical data with patients receiving strong CYP3A4 inhibitors, and/or inducers. Like temsirolimus, grapefruit and other foods known to inhibit CYP450 and PgP activity may increase exposure to everolimus and should be avoided during treatment[46]
.
ADRs and supportive care
mTOR inhibitors have a classically diverse side effect profile with several recommended management strategies. Established adverse event data in clinical trials for everolimus with more than 30% incidence across all FDA approved indications include stomatitis, infections, rash, fatigue, diarrhoea, oedema, abdominal pain, nausea, fever, asthenia, cough, headache and decreased appetite[46]
. Severe adverse effects (grades three and four), occurring in over 5% of study patients, were stomatitis, infection, fatigue and dyspnoea. Laboratory deviations are also common; serious (grade three and four) haemoglobinaemia, lymphocytopenia, hyperglycaemia, hypophosphataemia were reported in more than 10% of subjects. All patients receiving temsirolimus had all grade side effects; of these, 67% patients reported grades three and four severe side effects. Temsirolimus observed the following adverse events in more than 30% of patients: asthenia, oedema, mucositis, anorexia, nausea and rash. Severe side effects (grades three and four) included asthenia, pain, dyspnoea and rash. Temsirolimus also causes significant haematologic toxicity; in the clinical trials that garnered its FDA approval, 94% had haemoglobinaemia, 53% had lymphocytopenia, 19% had neutropenia, and 40% had thrombocytopenia. Grades three and four events related to hemoglobinemia, lymphocytopenia and neutropenia affected over 5% of subjects. In the event of haematologic toxicity (absolute neutrophil count <1000/mm3 , platelets < 75,000/mm3), temsirolimus should be held until resolution to grade two haematologic toxicity or less; product labelling should be followed for recommendations[42]
. This section highlights the major class related side effects: non-infectious pneumonitis, metabolic disturbances and mucosal toxicity.
| Table 2: Summary of management strategies for ADRs of mTOR inhibitors | ||
|---|---|---|
| Treatment | Dose adjustments | |
| *Based on CTCAE Criteria 4.0[46],[70],[71],[73] | ||
| Non-infectious pneumonitis | Grade 1: no intervention, increase monitoring. Grade 2: consider corticosteroid treatment, rule out infection. Grade 3: consider corticosteroid treatment, oxygen support required. Rule out infection. Grade 4: consider corticosteroid treatment, ventilatory support required. Rule out infection. | Grade 1: none. Grade 2: consider holding until resolution to grade 1. May restart at 50% of initial dose with potential to re-escalate dose if tolerated. If recovery does not occur within four weeks, stop mTOR inhibitor. Grade 3: stop mTOR inhibitor until resolution to grade 1. Dose reduce mTOR inhibitor upon restarting. If grade 3 toxicity recurs, consider discontinuing mTOR inhibitor permanently. Grade 4: discontinue mTOR inhibitor. |
| Metabolic disturbances | All grades: start appropriate therapy and monitor (ex. metformin, statin, insulin). Utilise recommendations from established practice guidelines. Target values: LDL cholesterol <190mg/dL, triglycerides <300mg/dL, fasting glucose (non-diabetic) <125mg/dL, f asting glucose (diabetic) <160mg/dL, random glucose (diabetic) <200mg/dL. Note: If triglycerides >500mg/dL, initiate triglyceride lowering therapy before targeting LDL. | Grades 1 or 2: none. Grade 3: temporarily hold mTOR inhibitor. Upon resolution to grade 2 or lower, restart mTOR inhibitor at a lower dose. Grade 4: discontinue mTOR inhibitor. |
| Stomatitis | Grade 1: saline or non-alcohol based mouth rinse several times a day. Grades 2, 3, or 4: topical analgesic treatment (e.g. benzocaine, lidocaine). May consider topical corticosteroids (e.g. triamcinolone, prednisolone). Notes: avoid alcoholic, hydrogen peroxide, iodine and thyme containing rinses. Rule out infection. If azole antifungal is given for infection, hold mTOR inhibitor because of drug-drug interaction. | Grade 1: none. Grade 2: temporarily hold mTOR inhibitor until resolution to grade 1, and resume same mTOR inhibitor dose. If stomatitis recurs at this grade, hold mTOR inhibitor again until resolution, and restart mTOR inhibitor at lower dose. Grade 3: hold mTOR inhibitor until resolution to grade 1 or lower and restart at reduced dose. Grade 4: discontinue mTOR inhibitor. |
Non-infectious pneumonitis
A serious and potentially fatal class effect of mTOR inhibitors is non-infectious pneumonitis. It is difficult to diagnose as patients may not be symptomatic, and infectious or cancer related causes should be ruled out. Respiratory symptoms are generally nonspecific, but may present as hypoxia, pleural effusion, cough, fever or dyspnoea. Non-infectious pneumonitis tends to occur more frequently with everolimus compared with temsirolimus[64]
. Clinical trials reported up to a 19% rate of occurrence when everolimus was used in combination with exemestane. The rate of all grades non-infectious pneumonitis of everolimus monotherapy for PNET and RCC was 17% and 14%, respectively; grades three and four pneumonitis rates ranged from 3–4% and 0–0.5%, respectively[46]
. The median time to onset for non-infectious pneumonitis with everolimus was 108 days (range 24–257 days)[65]
. There is some data to suggest that patients who develop everolimus-related pneumonitis have higher overall survival rates[66]
. Temsirolimus in combination with bevacizumab reported a pneumonitis rate of 4.8%, with the majority being grade one and two[67]
. In the trial that granted temsirolimus its FDA approval, only 2% of patients were diagnosed with interstitial lung disease. Temsirolimus monotherapy showed higher rates of radiographic identified pneumonitis at 29% compared with interferon; of the temsirolimus group, 31% of patients presented with symptoms. The probability of developing pneumonitis was 21% at 8 weeks of treatment, 31% at 16 weeks, and up to 45% at 13 months[68].
Intervention is recommended for patients who develop severe pneumonitis. Asymptomatic patients on everolimus with only radiographic findings of pneumonitis (grade one CTCAE criteria) do not require dose adjustments and may continue therapy, but should be closely monitored. Patients with symptoms that do not interrupt activities of daily living (ADL) may hold therapy; infection should also be excluded. Treatment with corticosteroids may also be considered until symptoms improve to grade one or lower and, upon resolution, everolimus should be restarted at a 50% dose reduction with potential to re-escalate the dose if tolerated. If symptoms do not resolve after four weeks, treatment should be permanently discontinued. Symptomatic patients who have issues completing ADL (i.e. grade three) should hold everolimus at least until resolution to grade one, and receive supportive oxygen and corticosteroids. Treatment with everolimus may then be restarted after two weeks at a 50% reduced dose. The same grade three precautions are recommended in life threatening, grade four pneumonitis in which ventilatory support is needed, and everolimus should be permanently discontinued[20],[46],[64]
. Temsirolimus does not have definitive treatment recommendations for non-infectious pneumonitis, but similar treatment strategies with antibiotics, steroids, dose reductions, or discontinuation of therapy were employed in phase III trials[69]
. General steroid dosing may be initiated with prednisone 0.75–1.0mg/kg or its equivalent. For more severe cases, methylprednisolone 2–5mg/kg/day in divided doses can be used initially. Duration of steroid therapy is varied based on time to symptom resolution and with longer treatment durations, tapering of steroids should be considered[70]
.
Metabolic disturbances
Metabolic changes are common but manageable side effects associated with mTOR inhibitors. Although hyperglycaemia and hyperlipidaemia caused by mTOR inhibitors are rarely acute medical issues, they can have serious sequelae if left untreated. The rapamycin pathway is linked with insulin signalling, and mTOR inhibitors are thought to cause insulin resistance and prevent lipid clearance from the bloodstream, resulting in increased levels of lipids and glucose[71]
. Obtaining a baseline fasting serum glucose and lipid panel is recommended prior to starting treatment and periodically while on therapy. Patients who already have hyperglycaemia and hyperlipidaemia should achieve target goals before starting on mTOR inhibitors and may require additional monitoring. In clinical trials of everolimus, metabolic disturbances caused significant increases in cholesterol, triglycerides and blood glucose. Hypercholesterolaemia of all grades affected around 75% of patients with solid tumours on everolimus, with up to 4% of patients experiencing grade three events. Triglycerides were elevated in 40–70%, with less than 1% above a grade two. Around 70% of patients developed impaired glucose, and of those only 9–17% were grade three[20],[46]
. Despite the high incidence of metabolic disturbances with everolimus, these disturbances were mostly mild to moderate in severity and there were no reported life threatening events. Studies with temsirolimus were similar with comparable prevalence of severe (grade three-four) adverse events. Hyperlipidaemia, hypercholesterolaemia and hyperglycaemia affected 27%, 24%, and 26% of patients, respectively. Grade three or four events were observed in 3% of patients with hyperlipidaemia, 11% with hyperglycaemia, and only 1% with hypercholesterolaemia[5]
.
Management strategies for metabolic disturbances follow current recommendations by established practice guidelines. mTOR inhibitors may be continued if only mild to moderate adverse effects are observed. Everolimus may be held for grade two events until resolution to grade one if side effects are intolerable. In severe grade three events, the dose should be held and, upon resolution of side effects, the mTOR inhibitor may be restarted at a lower dose (i.e. temsirolimus may be reduced by 5mg increments, but no lower than 15mg/day to maintain efficacy). Life threatening events warrant the permanent discontinuation of the drug[46]
. The National Cancer Institute has made recommendations based on expert opinion, with endorsements of the National Cholesterol Education Program (third report) and the European Society of Cardiology for the management of dyslipidaemia[72]
. The American Diabetes Association and the European Society for the Study of Diabetes Guidelines may be followed for the management of hyperglycaemia[73]
. Prior to starting mTOR inhibitors, a full lipid panel and glucose level (fasting or random) should be collected. Current target goals are as follows: fasting low density lipoprotein (LDL) cholesterol is <190mg/dL (lower goals may be warranted depending on cardiovascular risk), fasting triglycerides <300mg/dL, non-diabetic fasting blood glucose <125mg/dL, diabetic patient fasting blood glucose <160mg/dL, or random <200mg/dL. If life expectancy is estimated to be less than one year, the fasting triglyceride goal may be raised to <500mg/dL. Ideally, metabolic disturbances should be resolved prior to treatment, but starting treatment with management plans in place is feasible. The risk of pancreatitis increases when triglycerides greater than 500mg/dL; fibrates, extended release niacin, or omega 3 fatty acids are recommended in addition to therapeutic lifestyle changes (TLC) for the management elevated triglycerides. TLC (diet and exercise) for at least three months is recommended in all patients who are not at or below their LDL goal. For patients who do not reach goals, despite TLC, providers should consider starting statin therapy, optimising statin therapy, or adding another cholesterol lowering medication[46]
. Intermittent monitoring with a lipid panel at least once every cycle is necessary for any additions or modifications to treatment. When lipid values are stable, the taskforce recommends checking every three months[71]
.
Routine blood glucose monitoring is advised during each office visit. Diabetic and high risk diabetic patients should be counselled to initially increase their home glucose monitoring, particularly during the first week of starting an mTOR inhibitor; they may taper monitoring to a couple times per week for subsequent cycles of treatment if values are within their normal ranges. More frequent observation should be employed if patients are taking other drugs that cause hyperglycemia, such as steroids[46]
. Close to 90% of subjects in the temsirolimus phase III trial had at minimum one elevated glucose level on study, but only 26% were documented as an adverse event[42]
. Although hyperglycaemia is common, it does not always warrant intervention. If glucose levels are persistently mild to moderately elevated (grades one to three), oral antidiabetic agents should be initiated. Metformin is the initial drug of choice, barring any contraindications (e.g. hypersensitivity, renal dysfunction and lactic acidosis). For grade two hyperglycaemia, the task force initially recommends TLC and metformin, and if levels are still elevated after two weeks, a sulfonylurea may be added. If levels are still high a week later, basal insulin may be added to the two oral agents; continued hyperglycaemia beyond these interventions warrant stopping oral antidiabetic agents and starting an insulin treatment plan. Patients diagnosed with grade three or four hyperglycaemia should be treated more acutely: the mTOR inhibitor should be held, fluids should be given, and an aggressive insulin treatment plan should be initiated. Blood glucose monitoring at home is recommended in conjunction with insulin therapy. If glucose levels are still uncontrolled and consistently at levels over 250mg/dL, mTOR inhibitors should be held until resolution of the hyperglycaemic episode[71]
.
Mucosal toxicity
Stomatitis is the dose limiting toxicity associated with mTOR inhibitors. It is described as “aphthous-like” lesions, appearing as oval shaped, shallow ulcers with erythematous margins[74]
. A meta-analysis concluded that the onset of mucositis occurs early on in treatment, typically within the first week of starting an mTOR inhibitor[75]
. In clinical trials with everolimus, oral lesions were reported in 44–78% of patients, with grade three to four in 4–9% of these patients[46]
. Temsirolimus clinical trials noted a 41% incidence of all grade mucositis, and only 3% events were grade three and above. A small study by de Oliveira et al. found that 47% of patients required dose reductions or interruptions in therapy secondary to mucositis[76]
. Mild symptoms (grade one) that do not impair normal diet do not need an intervention. General oral hygiene should be employed. Frequent, non-medicated oral rinses (i.e. saline rinse, up to six times per day) can be used to reduce side effects, use of a soft toothbrush can decrease mechanical irritation, and the avoidance of alcohol or peroxide based mouth rinses are all good clinical practices[77]
. If patients experience moderate pain related to mucositis, consider holding mTOR inhibitors until mucositis resolves to grade one, and infectious causes should be ruled out. Upon resolution to grade one from grade two or three mucositis, the mTOR inhibitor should be restarted at a lower dose. Supportive care medications are also recommended; topical analgesics (e.g. lidocaine, benzocaine) with or without topical corticosteroids (e.g. triamcinolone, prednisolone) may be used to alleviate pain and irritation. Life threatening mucositis warrants the discontinuation of the mTOR inhibitor and treatment with appropriate medications and supportive care[46]
. If patients develop a viral or fungal oral infection, they should be treated with proper antimicrobials[78]
. If an azole antifungal (e.g. posaconazole or itraconazole) is prescribed, the mTOR inhibitor should be held on account of drug-drug interactions, which results in elevated serum levels of everolimus or temsirolimus, potentiating their toxic side effects[46]
.
Looking ahead
Mechanism of resistance to mTOR inhibitors
The pervasive nature of the mTOR pathway throughout various cell types lends itself to an assortment of mutations. Preclinical models have shown that heterogeneity within the mTOR pathway may direct a tumour’s sensitivity or resistance to mTOR inhibitors[79]
. Basic research findings also show that MTOR has the highest incidence of recurrent mutations located in six primary clusters that have phenotypic effects that can cause increased pathway activation[80]
. This makes the mTOR protein a primary target for inhibition and for the development of resistance. It is theorised that increased phosphorylation of cell signalling proteins such as AKT, S6K1 and 4EBP1 is an indication of an increased activation of PI3K/AKT/mTOR pathway, making it more sensitive to mTOR inhibitors. The degree of mTOR activation is commonly evaluated through the phosphorylation of S6K1, which is activated downstream from mTOR; decreased levels of phosphorylated AKT and S6K1 result in relative resistance to mTOR inhibitors[49]
. Resistance to mTOR inhibitors can occur via other mutated genes in the mTOR pathway. For example, the recurrent RHEB mutation can increase phosphorylation of S6K1 downstream causing increased resistance to mTORC1 inhibitors[80]
. Altering the negative feedback inhibition of the pathway through the addition of mTOR inhibitors can also promote acquired resistance. Blocking mTOR can induce activation of the AKT pathway through an IGF-IR (insulin-like growth factor-1 receptor) mechanism, thus lessening the efficacy of mTOR inhibitors[81],[82]
. Another proposed method of resistance is in the alteration of natural inhibitory binding sites on the mTOR protein itself. Certain mutations have been shown to decrease the binding potential of the endogenous inhibitor DEPTOR (DEP domain-containing mTOR-interacting protein) to mTOR, consequently allowing mTOR to maintain its downstream activity[80]
. The availability of resources that drive cell growth have also contributed to drug resistant mutations; mTORC1 is downregulated when there is a scarcity of amino acids and glucose, slowing down cell growth and proliferation. Certain mutated cells have been found to be circumvent this downregulation by constitutively activating SK61 and other signals downstream of mTOR, stimulating the cell to reproduce and grow despite the deprivation of resources. These various resistance mechanisms emphasise the need for the continued development of agents that are more resilient to mutations.
Future directions
Despite progression-free and overall survival benefit, mTOR inhibitors appear to be cytostatic rather than cytotoxic, as the majority of the patients treated with these inhibitors experience stable disease instead of complete remission. Unfortunately, over time patients will progress despite appropriate treatment with an mTOR inhibitor — most likely attributed to various resistance mechanisms. Resistance in cancer cells to mTOR inhibition highlights the need for multitargeted treatment strategies; several PI3K, mTORC1/2 and AKT inhibitors are currently under investigation with promising results in initial human trials, cancer cell lines and xenograft models[83]
.
mTORC1/2 inhibitors
Temsirolimus and everolimus only block one mTOR protein complex, mTORC1. Research reveals that the mTORC2 complex promotes AKT phosphorylation and may be linked to cell migration, resulting in metastasis[3]
. Nonselective mTORC1/2 inhibition may result in increased anticancer activity and better blockade of the PI3K/AKT cascade. Ongoing clinical trials of mTORC1/2 inhibitors are undergoing studies in gynaecological malignancies (ClinicalTrials.gov NCT02208375), lung cancer (ClinicalTrials.gov NCT02417701), breast cancer (ClinicalTrials.gov NCT02049957; NCT02619669), and liver cancer (ClinicalTrials.gov NCT02575339). MLN0128/INK128, an oral mTORC1/2 inhibitor, shows preliminary antitumour activity in human breast cancer cells with PIK3CA and PTEN mutations[84]
. MLN0128 also shows potent inhibitory effects on RCC growth and metastasis[85]
. CC-223 is another oral mTORC1/2 inhibitor that demonstrates preliminary antitumour activity in patients with advanced solid tumours or multiple myeloma in a phase I dose escalation study[86]
. Despite the theory that the inhibitory effect with a mTORC1/2 inhibitor is more potent, a randomised phase II study of AZD2014 versus everolimus in patients with VEGFR-refractory metastatic clear cell renal cancer found that the PFS and OS of AZD2014 is inferior to everolimus[87]
. However, there are ongoing phase II trials that continue to evaluate its potential benefit in lung cancer (Clinicaltrials.gov NCT02403895) and breast cancer (Clinicaltrials.gov NCT02216786).
PI3K/mTOR inhibitors
Dual PI3K/mTOR inhibitors are designed to target multiple areas of the pathway with the aim of achieving a synergistic inhibitory effect of the dysregulated pathway[88]
. A recent phase II study demonstrates modest activity by BEZ235 in patients with advanced transitional cell carcinoma; the median PFS was 62 days and median overall survival was 127 days. However, it has an unfavourable toxicity profile with 90% of the patients experiencing a drug-related event, and 50% of those patients had grade three or four adverse events[89]
. BEZ235 is also currently being studied in a phase II trial in patients with advanced PNET after failure of mTOR inhibitor therapy (ClinicalTrials.gov NCT01658436). Apitolisib (GDC-0980), a once-daily oral dual PI3K and mTOR inhibitor, has shown promising results in its first-in-human phase I trial. Dose-limiting toxicities included rash and hyperglycaemia. The most common treatment related adverse effects were hyperglycaemia, rash, elevated transaminases, diarrhoea, pneumonitis and mucosal inflammation[90]
. Currently, GDC-0980 is undergoing two phase II clinical trials (ClinicalTrials.gov NCT01455493; NCT01442090).
PI3K-selective inhibitors
Buparlisib (BKM120) is a once daily oral PI3K-selective inhibitor presently being studied in patients with advanced solid tumours. Results from the phase I, dose escalation study show that BKM120 is safe and well-tolerated with evidence of preliminary antitumour activity[91]
. One of the 31 patients with triple-negative breast cancer achieved a partial response. A total of 16 patients (52%) had stable disease for more than six weeks. Dose-limiting toxicities included mood alteration, hyperglycaemia, epigastralgia, and rash. Common adverse drug effects include rash, hyperglycaemia, diarrhoea, anorexia, and mood alterations. BKM120 is currently undergoing phase III trials in breast cancer (ClinicalTrials.gov NCT01610284; NCT01633060), and phase II trials in non-small cell lung cancer (ClincialTrials.gov NCT01487265 NCT01911325) and cervical cancer (ClincialTrials.gov NCT01613677).
AKT inhibitors
AKT activation leads to tumour angiogenesis and increased cell survival. MK-2206 is an oral pan-AKT inhibitor that demonstrates clinical activity in patients with advanced solid tumours. Adverse drug effects included skin rash, nausea, pruritus, hyperglycaemia and diarrhoea[92]
. MK2206 is currently in phase II trials for breast cancer (ClinicalTrials.gov NCT01277757), ovarian cancer (ClinicalTrials.gov NCT01283035), and refractory kidney cancer (ClinicalTrials.gov NCT01239342). Ipatasertib (GDC-0068), is an oral AKT inhibitor that blocks AKT signalling in cultured human cancer cell lines and tumour xenograft models[93],[94]
. Ipataseritib is undergoing investigation in patients with refractory solid tumors (ClinicalTrials.gov NCT01090960) and in breast cancer (ClinicalTrials.gov NCT02301988).
Conclusions
With the advent of genome sequencing and the growing understanding of oncogenes and cell signalling cascades, anticancer therapy is becoming more precise and personalised. The PI3K/AKT/mTOR signalling pathway plays a major role in cell proliferation, growth and survival, making it a popular area of cancer research. mTOR inhibitors have demonstrated improved PFS and OS in patients with advanced and metastatic disease. mTOR inhibitors have a unique class toxicity profile, which warrants attentive monitoring and management. Today, temsirolimus and everolimus are widely used in the treatment of breast cancer, RCC and pancreatic neuroendocrine tumors. The ubiquitous nature of the mTOR pathway allows these inhibitors to successfully target numerous disease states. However, the emergence of resistance to mTOR inhibitors has proven to be a challenge in maintaining durable responses in patients. Ongoing clinical research is investigating several pan-selective novel agents either as monotherapy or combination therapy in various solid malignancies. In the near future, treatment strategies of these advanced cancers will include cytotoxic agents, targeted therapy and immunotherapy in an attempt to control, and hopefully cure, such a resilient disease.
Tiffany Lin, PharmD, BCPS, BCOP is oncology pharmacist at Cedars-Sinai Medical Center. Crystal Leung, PharmD, BCPS, BCOP is oncology pharmacist at Cedars-Sinai Medical Center. Kieu Thuy Nguyen, PharmD is PGY2 oncology pharmacy resident at Cedars-Sinai Medical Center. Robert A. Figlin is Stephen Spielberg Family Chair in hematological oncology, professor of medicine and biomedical sciences, director, Division of Hematology Oncology, deputy director, Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Medical Center, 8700 Beverly Blvd., Los Angeles, CA 90048, Uniterd States. Correspondence to:
Tiffany.Lin@cshs.org
Financial and conflicts of interest disclosure:
The authors have no relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilised in the production of this manuscript.
Reading this article counts towards your CPD
You can use the following forms to record your learning and action points from this article from Pharmaceutical Journal Publications.
Your CPD module results are stored against your account here at The Pharmaceutical Journal. You must be registered and logged into the site to do this. To review your module results, go to the ‘My Account’ tab and then ‘My CPD’.
Any training, learning or development activities that you undertake for CPD can also be recorded as evidence as part of your RPS Faculty practice-based portfolio when preparing for Faculty membership. To start your RPS Faculty journey today, access the portfolio and tools at www.rpharms.com/Faculty
If your learning was planned in advance, please click:
If your learning was spontaneous, please click:
References
[1] Vivanco I & Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002;2:489–501. doi: 10.1038/nrc839
[2] Cargnello M, Tcherkezian J & Roux PP. The expanding role of mTOR in cancer cell growth and proliferation. Mutagenesis 2015;30:169–176. doi: 10.1093/mutage/geu045
[3] Meric-Bernstam F & Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol 2009;27:2278–2287. doi: 10.1200/JCO.2008.20.0766
[4] Galanis E, Buckner JC, Maurer MJ et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol 2005;23:5294–5304. doi: 10.1200/JCO.2005.23.622
[5] Hudes G, Carducci M, Tomczak P et al. Temsirolimus, Interferon Alfa, or Both for Advanced Renal-Cell Carcinoma. N Engl J Med 2007;356:2271–2281. doi: 10.1056/NEJMoa066838
[6] Andre F, Campone M, O’Regan R et al. Phase I study of everolimus plus weekly paclitaxel and trastuzumab in patients with metastatic breast cancer pretreated with trastuzumab. J Clin Oncol 2010;28:5110–5115. doi: 10.1200/JCO.2009.27.8549
[7] Oza AM, Elit L, Tsao MS et al. Phase II study of temsirolimus in women with recurrent or metastatic endometrial cancer: a trial of the NCIC Clinical Trials Group. J Clin Oncol 2011;29:3278–3285. doi: 10.1200/JCO.2010.34.1578
[8] Yao JC, Shah MH, Ito T et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011;364:514–523. doi: 10.1056/NEJMoa1009290
[9] Witzig TE, Geyer SM, Ghobrial I et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 2005;23:5347–5356. doi: 10.1200/JCO.2005.13.466
[10] Hess G, Herbrecht R, Romaguera J et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009;27:3822–3829. doi: 10.1200/JCO.2008.20.7977
[11] Smith SM, Van Besien K, Karrison T et al. Temsirolimus has activity in non-mantle cell non-Hodgkin’s lymphoma subtypes: The University of Chicago phase II consortium. J Clin Oncol 2010;28:4740–4746. doi: 10.1200/JCO.2010.29.2813
[12] Wang M, Popplewell LL, Collins RH et al. Everolimus for patients with mantle cell lymphoma refractory to or intolerant of bortezomib: multicentre, single-arm, phase 2 study. Br J Haematol 2014;165:510–518. doi: 10.1111/bjh.12780
[13] Ghobrial IM, Witzig TE, Gertz M et al. Long-term results of the phase II trial of the oral mTOR inhibitor everolimus (RAD001) in relapsed or refractory Waldenstrom Macroglobulinemia. Am J Hematol 2014;89:237–242. doi: 10.1002/ajh.23620
[14] Bukowski RM, Negrier S & Elson P. Prognostic factors in patients with advanced renal cell carcinoma: development of an international kidney cancer working group. Clin Cancer Res 2004;10:6310S–6314S. doi: 10.1158/1078-0432.CCR-050000
[15] Motzer RJ, Bukowski R, Figlin R et al. Prognostic nomogram for sunitinib in patients with metastatic renal cell carcinoma. Cancer 2008;113:1552–1558. doi: 10.1002/cncr.23776
[16] Motzer RJ, Jonasch E, Agarwal N et al. Kidney cancer, version 3. 2015. J Natl Compr Canc Netw 2015;13:151–159. PMID: 25691606
[17] Escudier B, Porta C, Schmidinger M et al. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25:iii49–56. doi: 10.1093/annonc/mdu259
[18] De Reijke TM, Bellmunt J, Van Poppel H et al. EORTC-GU group expert opinion on metastatic renal cell cancer. Eur J Cancer 2009;45:765–773. doi: 10.1016/j.ejca.2008.12.010
[19] Ljungberg B, Bensalah K, Canfield S et al. EAU guidelines on renal cell carcinoma: 2014 update. Eur Urol 2015;67:913–924. doi: 10.1016/j.eururo.2015.01.005
[20] Motzer RJ, Escudier B, Oudard S et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. The Lancet 2008;372:449–456. doi: 10.1016/S0140-6736(08)61039-9
[21] Motzer RJ, Barrios CH, Kim TM et al. Phase II randomized trial comparing sequential first-line everolimus and second-line sunitinib versus first-line sunitinib and second-line everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol 2014;32:2765–2772. doi: 10.1200/JCO.2013.54.6911
[22] NICE. Everolimus for the second-line treatment of advanced renal cell carcinoma 1-guidance. Guidance and guidelines, 2011. Available at: http://www.nice.org.uk/guidance/TA219/chapter/1-Guidance (accessed 24 February 2016).
[23] Motzer RJ, Hutson TE, Glen H et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. The Lancet Oncol 2015. doi: 10.1016/S1470-2045(15)00290-9
[24] Choueiri TK, Escudier B, Powles T et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015. doi: 10.1056/NEJMoa1510016
[25] Motzer RJ, Escudier B, Mcdermott DF et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015. doi: 10.1056/NEJMoa1510665
[26] Zagouri F, Sergentanis TN, Chrysikos D et al. mTOR inhibitors in breast cancer: a systematic review. Gynecol Oncol 2012;127:662–672. doi: 10.1016/j.ygyno.2012.08.040
[27] Dorris JR & Jones S. Everolimus in Breast Cancer: The Role of the Pharmacist. Ann Pharmacother 2014;48:1194–1201. doi: 10.1177/1060028014542415
[28] Shtivelband MI. Everolimus in hormone receptor-positive advanced breast cancer: targeting receptor-based mechanisms of resistance. Breast 2013;(4):405–410. doi: 10.1016/j.breast.2013.02.003
[29] Berns K, Horling HM, Hennessy BT et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030
[30] Nagata Y, Lan KH, Zhou X et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004;6:117–127. doi: 10.1016/j.ccr.2004.06.022
[31] Esteva FJ, Guo H, Zhang S et al. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol 2010;177(4):1647–1656. doi: 10.2353/ajpath.2010.090885
[32] Morrow PK, Wulf GM, Ensor J et al. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol 2011;29:3126–3132. doi: 10.1200/JCO.2010.32.2321
[33] Baselga J, Campone M, Piccart M et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 2012;366:520–529. doi: 10.1056/NEJMoa1109653
[34] Yardley DA, Noguchi S, Pritchard KI et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Avd Ther 2013;30:870–884. doi: 10.1007/s12325-013-0060-1
[35] Gradishar WJ, Anderson BO, Balassanian R et al. Breast Cancer, version 1.2016. J Natl Compr Canc Netw 2015;13:1475–1485. PMID: 26656517
[36] NICE. Everolimus in combination with exemestane for treating advanced HER2-negative hormone-receptor-positive breast cancer after endocrine therapy. Guidance and guidelines, 2013. Available at: http://www.nice.org.uk/guidance/TA295 (accessed 26 February 2016).
[37] Scottish Medicines Consortium. Everolimus (Afinitor), 2016. Available at: https://www.scottishmedicines.org.uk/SMC_Advice/Advice/872_13_everolimus_Afinitor/everolimus_Afinitor_RE-SUBMISSION (accessed 26 February 2016).
[38] Yao JC. Neuroendocrine tumors. Molecular targeted therapy for carcinoid and islet-cell carcinoma. Best Pract Res Clin Endocrinol Metab 2007;21:163–172. doi: 10.1016/j.beem.2007.01.006
[39] Von wichert G, Jehle PM, Hoeflich A et al. Insulin-like growth factor-I is an autocrine regulator of chromogranin A secretion and growth in human neuroendocrine tumor cells. Cancer Res 2000;60:4573–4581. PMID: 10969809
[40] Pavel ME, Hainsworth JD, Baudin E et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. The Lancet 2011;378:2005–2012. doi: 10.1016/S0140-6736(11)61742-X
[41] FROM ECC 2015-neuroendocrine cancer: RADIANT-4 trial-NET improvement with everolimus? Nat Rev Clin Oncol 2015. doi: 10.1038/nrclinonc.2015.181
[42] Torisel [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals, Inc; 2007.
[43] European Medicines Agency. Torisel, 2016. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000799/human_med_001098.jsp&mid=WC0b01ac058001d124 (accessed 25 February 2016).
[44] Raymond E, Alexandre J, Faivre S et al. Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. J Clin Oncol 2004;22:2336–2347. doi: 10.1200/JCO.2004.08.116
[45] Hidalgo M, Buckner JC, Erlichman C et al. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res 2006;12:5755–5763. doi: 10.1158/1078-0432.CCR-06-0118
[46] Afinitor [package insert]. East Hanover, NJ: Novartis Pharmaceuticals, Corp; 2008.
[47] Beuvink I, Boulay A, Fumagalli S et al. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell 2005;120:747–759. doi: 10.1016/j.cell.2004.12.040
[48] Negrier S, Gravis G, Perol D et al. Temsirolimus and bevacizumab, or sunitinib, or interferon alfa and bevacizumab for patients with advanced renal cell carcinoma (TORAVA): a randomised phase 2 trial. The Lancet Oncol 2011 Jul;12:673–680. doi: 10.1016/S1470-2045(11)70124-3
[49] Faivre S, Kroemer G & Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov 2006;5:671–688. doi: 10.1038/nrd2062
[50] Boni JP, Hug B, Leister C et al. Intravenous temsirolimus in cancer patients: clinical pharmacology and dosing considerations. Semin Oncol 2009;36:S18–25. doi: 10.1053/j.seminoncol.2009.10.009
[51] Kovarik JM, Noe A, Berthier S et al. Clinical development of an everolimus pediatric formulation: relative bioavailability, food effect, and steady-state pharmacokinetics. J Clin Pharmacol 2003;43:141–147. doi: 10.1177/0091270002239822
[52] Kovarik JM, Hartmann S, Figueiredo J et al. Effect of food on everolimus absorption: quantification in healthy subjects and a confirmatory screening in patients with renal transplants. Pharmacotherapy 2002;22:154–159. doi: 10.1592/phco.22.3.154.33542
[53] Lunardi G, Armirotti A, Nicodemo M et al. Comparison of temsirolimus pharmacokinetics in patients with renal cell carcinoma not receiving dialysis and those receiving hemodialysis: a case series. Clin Ther 2009;31:1812–1819. doi: 10.1016/j.clinthera.2009.08.018
[54] Lunardi G, Vannozzi MO, Armirotti A et al. Temsirolimus in patients with renal cancer on hemodialysis. J Clin Oncol 2008;26:5652–5653. doi: 10.1200/JCO.2008.19.3144
[55] Sarantopoulos J, Lenz H, LoRusso P et al. Phase I pharmacokinetic study of temsirolimus (CCI-779) in patients with advanced malignancies and normal and impaired liver function: An NCI Organ Dysfunction Working Group (ODWG) study; 2011. American Society of Clinical Oncology Annual Meeting; 2011. Chicago, IL. J Clin Oncol 29:2011 (suppl; abstr 3072).
[56] Peveling-Oberhag J, Zeuzem S, Yong WP et al. Effects of hepatic impairment on the pharmacokinetics of everolimus: a single-dose, open-label, parallel-group study. Clin Ther 2013;35:215–225. doi: 10.1016/j.clinthera.2013.02.007
[57] Kovarik JM, Hsu CH, McMahon L et al. Population pharmacokinetics of everolimus in de novo renal transplant patients: impact of ethnicity and comedications. Clin Pharmacol Ther 2001;70:247–254. doi: 10.1067/mcp.2001.118022
[58] Prichard KI, Burris HA III, Ito Y et al. Safety and efficacy of everolimus with exemestane vs. exemestane alone in elderly patients with HER2-negative, hormone receptor-positive breast cancer in BOLERO-2. Clin Breast Cancer 2013;13:421–432. e1–e8. doi: 10.1016/j.clbc.2013.08.011
[59] Fujisaka Y, Yamada Y, Yamamoto N et al. A Phase 1 clinical study of temsirolimus (CCI-779) in Japanese patients with advanced solid tumors. Jpn J Clin Oncol 2010;40:732–738. doi: 10.1093/jjco/hyq047
[60] Boni JP, Leister C, Burns J et al. Differential effects of ketoconazole on exposure to temsirolimus following intravenous infusion of temsirolimus. Br J Cancer 2008;98:1797–1802. doi: 10.1038/sj.bjc.6604376
[61] Boni J, Leister C, Burns J et al. Pharmacokinetic profile of temsirolimus with concomitant administration of cytochrome p450-inducing medications. J Clin Pharmacol 2007;47:1430–1439. doi: 10.1177/0091270007306957
[62] Kovarik JM, Hartmann S, Hubert M et al. Pharmacokinetic and pharmacodynamic assessments of HMG-CoA reductase inhibitors when coadministered with everolimus. J Clin Pharmacol 2002;42:222–228. doi: 00912700222011148
[63] Grabowsky JA. Drug interactions and the pharmacist: focus on everolimus. Ann Pharmacother 2013;47:1055–1063. doi: 10.1345/aph.1R769
[64] Nozawa M, Ohzeki T, Tamada S et al. Differences in adverse event profiles between everolimus and temsirolimus and the risk factors for non-infectious pneumonitis in advanced renal cell carcinoma. Int J Clin Oncol 2015;20:790–795. doi: 10.1007/s10147-014-0764-5
[65] Ravaud A. Treatment-Associated Adverse Event Management in the Advanced Renal Cell Carcinoma Patient Treated with Targeted Therapies. Oncologist 2011;16:32–44. doi: 10.1634/theoncologist.2011-S2-32
[66] Cauley DH, Atkinson BJ, Ng CS et al. Everolimus (E) and temsirolimus (T) associated pneumonitis (P) in patients with metastatic renal cell cancer (mRCC): A single- center experience. J Clin Oncol 2012:suppl 5; abstr 401.
[67] Rini BI, Bellmunt J, Clancy J et al. Randomized phase III trial of temsirolimus and bevacizumab versus interferon alfa and bevacizumab in metastatic renal cell carcinoma: INTORACT trial. J Clin Oncol 2014;32:752–759. doi: 10.1200/JCO.2013.50.5305
[68] Maroto JP, Hudes G, Dutcher JP et al. Drug-related pneumonitis in patients with advanced renal cell carcinoma treated with temsirolimus. J Clin Oncol 2011;29:1750–1756. doi: 10.1200/JCO.2010.29.2235
[69] Bellmunt J, Szczylik C, Feingold J et al. Temsirolimus safety profile and management of toxic effects in patients with advanced renal cell carcinoma and poor prognostic features. Ann Oncol 2008;19:1387–1392. doi: 10.1093/annonc/mdn066
[70] Albiges L, Chamming’s F, Duclos B et al. Incidence and management of mTOR inhibitor-associated pneumonitis in patients with metastatic renal cell carcinoma. Ann Oncol 2012;23:1943–1953. doi: 10.1093/annonc/mds115
[71] Busaidy NL, Farooki A, Dowlati A et al. Management of metabolic effects associated with anticancer agents targeting the PI3K-AKT-mTOR pathway. J Clin Oncol 2012;30:2919–2928. doi: 10.1200/JCO.2011.39.7356
[72] National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002;106:3143–3421. PMID: 12485966
[73] Inzucchi SE, Bergenstal RM, Buse JB et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015;38:140–149. doi: 10.2337/dc14-2441
[74] Sonis S, Treister N, Chawla S et al. Preliminary characterization of oral lesions associated with inhibitors of mammalian target of rapamycin in cancer patients. Cancer 2010;116:210–215. doi: 10.1002/cncr.24696
[75] Martins F, de Oliveira MA, Wang Q et al. A review of oral toxicity associated with mTOR inhibitor therapy in cancer patients. Oral Oncol 2013;49:293–298. doi: 10.1016/j.oraloncology.2012.11.008
[76] de Oliveira MA, Martins E Martins F, Wang Q et al. Clinical presentation and management of mTOR inhibitor-associated stomatitis. Oral Oncol 2011;47:998–1003. doi: 10.1016/j.oraloncology.2011.08.009
[77] Peterson DE, Bensadoun RJ & Roila F. Management of oral and gastrointestinal mucositis: ESMO Clinical Practice Guidelines. Ann Oncol 2011;22:vi78–84. doi: 10.1093/annonc/mdr391
[78] Porta C, Osanto S, Ravaud A et al. Management of adverse events associated with the use of everolimus in patients with advanced renal cell carcinoma. Eur J Cancer 2011;47:1287–1298. doi: 10.1016/j.ejca.2011.02.014
[79] Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol 2010;7:209–219. doi: 10.1038/nrclinonc.2010.21
[80] Grabiner BC, Nardi V, Birsoy K et al. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov 2014;4:554–563. doi: 10.1158/2159-8290.CD-13-0929
[81] O’Reilly KE, Rojo F, She QB et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925
[82] Wan X, Harkavy B, Shen N et al. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007;26:1932–1940. doi: 10.1038/sj.onc.1209990
[83] Dienstmann R, Rodon J, Serra V et al. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther 2014;13:1021–1031. doi: 10.1158/1535-7163.MCT-13-0639
[84] Gökmen-Polar Y, Liu Y, Toroni RAet al. Investigational drug MLN0128, a novel TORC1/2 inhibitor, demonstrates potent oral antitumor activity in human breast cancer xenograft models. Breast Cancer Res Treat 2012;136:673–682. doi: 10.1007/s10549-012-2298-8
[85] Ingels A, Zhao H, Thong AE et al. Preclinical trial of a new dual mTOR inhibitor, MLN0128, using renal cell carcinoma tumorgrafts. Int J Cancer 2014;134:2322–2329. doi: 10.1002/ijc.28579
[86] Bendell JC, Kelley RK, Shih KC et al. A phase I dose-escalation study to assess safety, tolerability, pharmacokinetics, and preliminary efficacy of the dual mTORC1/mTORC2 kinase inhibitor CC-223 in patients with advanced solid tumors or multiple myeloma. Cancer 2015;121:3481–3490. doi: 10.1002/cncr.29422
[87] Powles T, Wheater M, Din O et al. A Randomised Phase 2 Study of AZD2014 Versus Everolimus in Patients with VEGF-Refractory Metastatic Clear Cell Renal Cancer. Eur Urol 2015. doi: 10.1016/j.eururo.2015.08.035
[88] Serra V, Markman B, Scaltriti M et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res 2008;68:8022–8030. doi: 10.1158/0008-5472.CAN-08-1385
[89] Seront E, Rottey S, Filleul B et al. Phase II study of dual phosphoinositol-3-kinase (PI3K) and mammalian target of rapamycin (mTOR) inhibitor BEZ235 in patients with locally advanced or metastatic transitional cell carcinoma (TCC). BJU Int 2016. doi: 10.1111/bju.13415
[90] Dolly S, Wagner AJ, Bendell JC et al. Phase I study of apitolisib (GDC-0980), dual phosphatidylinositol-3-kinase and mammalian target of rapamycin kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2016. doi: 10.1158/1078-0432.CCR-15-2225
[91] Bendell JC, Rodon J, Burris HA et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 2012;30:282–290. doi: 10.1200/JCO.2011.36.1360
[92] Yap TA, Yan L, Patnaik A et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol 2011;29:4688–4695. doi: 10.1200/JCO.2011.35.5263
[93] Lin J, Sampath D, Nannini MA et al. Targeting activated AKT with GDC-0068, a novel selective AKT inhibitor that is efficacious in multiple tumor models. Clin Cancer Res 2013;19:1760–1772. doi: 10.1158/1078-0432.CCR-12-3072
[94] Yan Y, Wagle M, Punnoose E et al. A first-in-human trial of GDC-0068: A novel, oral, ATP-competitive AKT inhibitor, demonstrates robust suppression of the AKT pathway in surrogate and tumor tissues. Mol Cancer Ther 2011;10:B154. doi: 10.1158/1535-7163.TARG-11-B154