

Biophoto Associates / Science Photo Library
Chronic Kidney Disease (CKD) is an international public health problem affecting 5–10% of the world population[1]
and is defined as a reduction in kidney function for at least three months.
Chronic Kidney Disease – Mineral and Bone Disorder (CKD-MBD) is used to describe the pathophysiological changes that occur in the vascular and skeletal system in CKD and has replaced the previous terminology — renal osteodystrophy and renal bone disease — in clinical practice[2]
.
The development of CKD-MBD starts early in the course of CKD on account of changes in the levels of parathyroid hormone (PTH), active vitamin D (calcitriol), and serum calcium and phosphate[1],
[2],
[3],
[4]
. Bone abnormalities are found in almost all patients on dialysis and in the majority (50–100%) of patients with CKD stage 3–5[1],
[3]
.
Research over the past few decades has uncovered many of the mechanisms involved in CKD-MBD. Emphasis is now placed on the need to start therapy early in the course of CKD. This article will outline the main mechanisms involved in CKD-MBD and the therapeutic interventions that aim to control this complication.
Pathophysiology
In normal bone, the remodelling process is tightly controlled. Osteoblasts produce a bone matrix from collagen and ground substances that become mineralised. Osteoclasts degrade bone to initiate normal bone remodelling and mediate bone loss in pathologic conditions by increasing their resorptive activity.
Normal parathyroid hormone regulation
The four parathyroid glands are located in the neck. Their function is to produce parathyroid hormone (PTH), which is secreted depending on calcium-sensing receptors and vitamin D receptors.
The main actions of PTH (see ‘ Normal parathyroid regulation in calcium and phosphate homeostasis’) are to increase serum calcium and decrease phosphate to maintain normal serum levels via three main mechanisms:
- Kidney: PTH blocks the reabsorption of phosphate at the proximal tubule causing increased excretion and also increases active reabsorption of calcium from the distal tubule. PTH stimulates 1α-hydroxylase activity to hydroxylate 25-hydroxyvitamin D forming active vitamin D (calcitriol).
- Bone: PTH increases the breakdown of bone mineral and collagen via cytokine-mediated osteoclast activity. PTH also promotes bone formation via direct action on osteoblasts.
- Gastrointestinal tract: the increased calcitriol produced by the kidney leads to increased gastrointestinal absorption of calcium and phosphate.
High calcium and active vitamin D levels act as negative feedback on the parathyroid gland and therefore homeostasis is achieved.
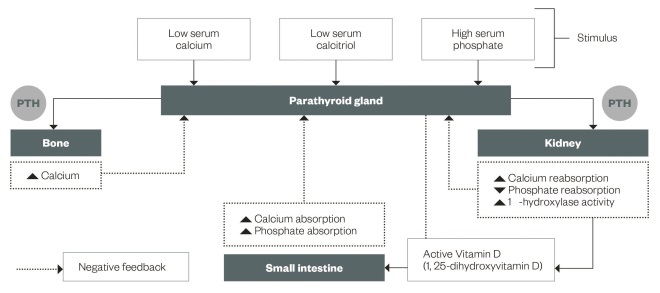
Figure 1: Normal parathyroid regulation in calcium and phosphate homeostasis
The main actions of PTH are to increase serum calcium and decrease phosphate to maintain normal serum levels via the kidney, bone and gastrointestinal tract.
Effect of CKD on calcium and phosphate homeostasis and PTH
As early as stage 3 CKD (estimated glomerular filtration rate [eGFR] 30–59ml/min/1.73m2) the body’s ability to excrete a phosphate load is diminished. This leads to phosphate retention causing a rise in PTH[5]
and a decline in calcitriol levels[6]
. The PTH rise causes reduced phosphate reabsorption and this compensatory hyperparathyroidism keeps serum phosphate levels within the normal range until kidney function declines further to stage 4 CKD (eGFR 15–29ml/min) when the kidneys’ ability to excrete phosphate is limited.
Serum calcitriol levels drop as eGFR falls below 40ml/min[3]
because of reduced renal calcitriol production and suppression of production by hyperphosphataemia[5]
. Lack of calcitriol and hyperphosphatemia leads to hypocalcaemia. Hypocalcaemia further stimulates PTH secretion via the calcium-sensing receptors, which can cause a PTH rise[7]
. However, the kidney fails to respond to prolonged raised levels of PTH. Long-term reduction of extracellular calcium over weeks or months promotes the development of parathyroid gland hyperplasia[7]
. The prolonged raised PTH is known as secondary hyperparathyroidism, since it is caused secondary to the effects of CKD. If the parathyroid glands become hyperplastic there is a decreased expression of calcium-sensing receptors[8]
, which means further increased PTH secretion caused by a lack of response to serum calcium. With advancing renal failure, and increasing uraemia, there is “skeletal resistance” to the effects of PTH[3],
[6]
. To achieve active bone turnover in these patients, maintaining higher levels of PTH compared to the normal population has been suggested to avoid adynamic bone disease[9]
(see ‘Renal Association recommended target ranges for bone parameters in Chronic Kidney Disease stages’).
| Renal Association recommended target ranges for bone parameters in Chronic Kidney Disease stages | |||
|---|---|---|---|
| Normal range | Renal Association range CKD stage 3–5 | Renal Association range CKD stage 5 dialysis | |
| Phosphate (mmol/L) | 0.7–1.4 | 0.9–1.5 | 1.1–1.7 |
| Adjusted calcium (mmol/L) | 2.2–2.6 | 2.2–2.6 (within the normal reference range) | 2.2–2.5 |
| Parathyroid hormone (pmol/L) | 1.6–9.9 | If consistently above normal range and rapidly rising, consider treatment | 2.0–9.0 times upper limit normal |
Fibroblast Growth Factor-23
In recent years there has been interest in the discovery of a new protein that is secreted by osteocytes; Fibroblast Growth Factor (FGF)-23. FGF-23 increases renal excretion of phosphate, reduces the kidneys’ 1α-hydroxylase enzyme activity and increases the catabolism of calcitriol[1],
[10],
[11]
. High levels of FGF-23 are seen in those with CKD and are thought to be protective against the initial phosphate rise in early CKD[1],
[10],
[11]
. Increased FGF-23 levels have been associated with mortality[12],
[13]
. As calcitriol increases FGF-23 levels this may lead to suggestions that vitamin D-sparing regimes are beneficial in reducing mortality from CKD-MBD. The rise in FGF-23 may be one of the early biomarkers for CKD-MBD, allowing for early intervention, but there is still much research required[10],
[11],
[12]
.
There are a number of types of bone disease seen in CKD-MBD[1],
[3],
[4],
[5]
:
- High bone turnover related to hyperparathyroidism;
- Mixed renal osteodystrophy with hyperparathyroidism and mineralisation defects;
- Low bone turnover, also known as adynamic bone disease, is a consequence of PTH over-suppression, vitamin D deficiency and aluminium deposition (the latter being rare because of the reduced use of aluminium-based phosphate binders);
- Some patients may also have superimposed osteoporosis.
In addition, metabolic acidosis is a common complication of CKD and can lead to increased calcium efflux from bone[4]
. This occurs because of bone buffering of hydrogen ions and an increase in bone resorption with reduced bone formation[4]
.
Diagnosis and clinical features
CKD-MBD may manifest as any of the following[1]
:
- Abnormalities of calcium, phosphorous, PTH and vitamin D metabolism;
- Abnormalities of bone turnover, mineralisation, volume and strength;
- Vascular or soft tissue calcification.
Bone biopsy would be the gold standard for the definitive diagnosis of CKD-MBD; however, this is not carried out in routine clinical practice because of its invasive nature and so the diagnosis is based on biochemical parameters[6]
.
PTH levels can be used as a marker for bone disease but are highly variable and so multiple measurements and trends need to be considered. PTH levels >50pmol/L are indicative of high bone turnover disease, whereas adynamic bone disease is suspected when PTH <10pmol/L.
Early bone disease with CKD is usually asymptomatic. Musculoskeletal symptoms appear late and include bone pain in the lower back, hips and legs that is aggravated by weight-bearing. Proximal muscle weakness may also be seen. A significant fracture risk is present with increasing PTH levels. Similar symptoms may be seen in both high and low bone turnover states[6]
.
Calciphylaxis is a serious complication of secondary hyperparathyroidism. It causes soft tissue calcification of small arterioles and venules that can lead to thrombosis and skin necrosis. Patients with calciphylaxis may experience excruciating pain and require large amounts of pain relief. Mortality is high on account of secondary infection and sepsis. Calcium and vitamin D products should be avoided in calciphylaxis.
Treatment
The aim of treatment includes maintaining serum levels of calcium and phosphate as near to normal as possible and preventing development of severe hyperparathyroidism and extraskeletal calcification, which may reduce fracture risk and cardiovascular disease. Treatment is usually started when patients reach stage 4 CKD and are in the care of specialist nephrology services.
Treatment of hyperphosphataemia
The National Institute for Health and Care Excellence (NICE) guidelines for hyperphosphataemia in CKD[14]
follow the Renal Association recommendations for target phosphate levels (see ‘Renal Association recommended target ranges for bone parameters in Chronic Kidney Disease stages’). However there are no randomised controlled trials that show the benefits of phosphate-lowering on patient survival. There is also no evidence that lowering phosphate to a particular level improves the clinical outcome, so suggested target levels are based on observational studies[9]
. Options for reducing phosphate should be a combination of pharmacological and non-pharmacological treatment[9],
[14]
.
Dietary management usually follows a referral to a renal dietitian, which is recommended to ensure that a reduced dietary phosphate intake is achieved while still maintaining overall nutritional adequacy of the diet. Advice can be given in relation to checking labels for phosphate additives and the renal dietitian can provide patients with written information to advise them on how to reduce the overall phosphate content of their diet. They can also play a role in supporting patients with phosphate binder compliance and how to tailor their medication to their meal pattern and phosphate content of food.
The main dietary sources of phosphate are:
- High-protein foods: foods that contain protein, such as milk, cheese, yoghurt and eggs, are the richest source of phosphate. Achieving a low dietary phosphate intake should not compromise overall nutritional status.
- Phosphate additives: manufacturers are increasingly adding phosphate in the form of phosphate additives to many different foods. Nearly half of our daily phosphate intake comes from phosphate additives and almost all of this is absorbed by the body. Phosphate additives therefore can have a greater effect on the levels in blood than phosphate from natural sources.
Phosphate binder therapy should be individualised for each patient, since evidence to suggest that any specific phosphate binder impacts upon patient outcome is lacking[9]
. There is some evidence that increased calcium load can increase vascular calcification and arterial stiffness but trials of calcium versus non-calcium binders have not shown significant differences in mortality[9]
. A meta-analysis of the effect of calcium versus non-calcium binders on mortality demonstrated a 22% reduction in all-cause mortality in those on non-calcium binders[15]
. However, these are observational studies and do not define causality.
Phosphate binders work by binding to dietary phosphate in the small intestine, forming insoluble phosphate compounds that reduce its absorption. The cations calcium, magnesium and lanthanum are able to bind phosphate anions forming insoluble phosphate salts. Sevelamer is a polymer with protonated amines that bind the phosphate anion. A new iron-based phosphate binder (Velphoro) binds phosphate in exchange for one of its hydroxyl groups.
Binders vary in their effectiveness in reducing phosphate absorption, which may affect the number of phosphate-binding tablets required. A study looking at phosphate-binding capacity of different binders found that lanthanum 1g (one tablet) ≡ calcium acetate 2g (two tablets) ≡ sevelamer 2.4g (three tablets)[16]
.
The suggested first line phosphate binder is calcium acetate[14]
. It has a reduced calcium load compared with calcium carbonate, and reducing long-term calcium load would seem reasonable[1],
[9]
. Calcium carbonate may be considered if the patient finds calcium acetate unpalatable or if the patient is hypocalcaemic.
If the patient is hypercalcaemic or hypoparathyroid then switching to, or starting with, a non-calcium based phosphate binder (e.g. sevelamer and lanthanum) should be undertaken (see ‘Available phosphate binders’).
| Available phosphate binders | ||
|---|---|---|
| Binder composition | Brands available | Comments |
| Calcium acetate | Renacet, Phosex, PhosLo | Usual first line binder |
| Calcium carbonate | Calcichew, Adcal | Avoid unless hypocalcaemic |
| Lanthanum carbonate | Fosrenol | |
| Sevelamer carbonate | Renvela, Generic by Consilient Health | |
| Sevelamer hydrochloride | Renagel | The carbonate salt is favoured due to the potential absorption of hydrochloride, exacerbating metabolic acidosis, seen in CKD |
| Calcium acetate/magnesium carbonate | Osvaren | May be used if calcium acetate is not tolerated before initiation of more expensive non-calcium binders |
| Aluminium hydroxide | AluCaps | Usually reserved for short-term use in difficult to treat patients |
| Sucroferric oxyhydroxide | Velphoro | Relatively new to the UK market |
| Ferric citrate | Ferexic | Approved by the European Medicines Agency in October 2015 |
Aluminium was first used as a phosphate binder in the 1940s. Its use has now decreased with the availability of other non-calcium-based binders, although some units may use short courses. Long-term use of aluminium binders led to concerns with its accumulation in renal disease[17]
and complications such as adynamic bone disease, microcytic anaemia, cardiovascular disease and dementia.
The main side effects of the binders are gastrointestinal disturbances, which include constipation, diarrhoea, nausea, vomiting and abdominal cramps. These occur in more than 10% of patients with lanthanum and sevelamer[18],
[19]
and around 1% of patients on calcium acetate[20]
. Sevelamer may also bind to bile acids, which could lead to reduced levels of fat-soluble vitamins.
Interactions with phosphate binders involve the reduced absorption of other drugs. The main interactions include:
- Levothyroxine (avoid lanthanum within two hours) with monitoring of thyroid function tests with all binders;
- Doxycycline (avoid lanthanum within two hours, avoid calcium acetate within three hours);
- Ciprofloxacin bioavailability is reduced by 50% when administered with binders (avoid lanthanum within two hours, avoid calcium acetate within three hours, avoid concomitant use of sevelamer).
Counselling points with phosphate binders
- Must be taken with meals to be effective.
- Calcium carbonate and lanthanum tablets must be chewed. Lanthanum tablets may not be suitable for patients with dentures.
- Sachets of lanthanum are available to sprinkle on food.
- Calcium acetate and sevelamer must be swallowed whole.
- Sevelamer sachets must be dispersed in 60ml of water. If this is being taken three times daily, this may have an impact on the amount of fluid the patient can take in, if on a strict fluid restriction.
- Check adherence as palatability can be problematic.
- Patients may split their binders up based on phosphate in food; for example, six binders per day is not a rigid two binders three times daily but can be four binders with a large phosphate meal, and one with each of the other meals.
Patients on long-hours home haemodialysis (>6 hours alternate days) remove significantly more phosphate than those on unit-based dialysis (four hours three times weekly). This may negate the need for phosphate-binding medication in patients on long-hours dialysis, with a relaxed diet avoiding phosphate restriction[21]
. It may even lead to hypophosphataemia and these patients should be encouraged to increase their dietary phosphate.
Treatment of hyperparathyroidism
Vitamin D3 is formed primarily following conversion of 7-dehydroxycholesterol in the skin through ultraviolet radiation. This needs to be hydroxylated in the liver to 25 OH vitamin D3 before finally being activated in the kidney by 1α-hydroxylase to 1, 25-dihydroxy vitamin D3 (calcitriol). This final process is reduced in stage 4-5 CKD because of reduced activity of this enzyme and so the use of activated vitamin is used to treat hyperparathyroidism.
There are two forms of activated vitamin D that are used in practice; calcitriol and hydroxyvitamin D3 (alfacalcidol). These products are equivalent in their efficacy for reducing PTH[22],
[23]
; however, if a patient had both liver and renal disease then calcitriol would be the treatment of choice because 1α-hydroxy vitamin D3 still requires hepatic hydroxylation.
To treat stage 4–5 CKD patients who are hypocalcaemic, activated vitamin D3 will be prescribed daily to optimise calcium absorption. The dose will be titrated depending on calcium levels and the usual dose will be 0.25–1µg daily. In acute deficiency states, up to 5µg daily may be used.
For patients who are normocalcaemic, or have an adjusted calcium level at the higher end of normal range, then “pulsed” activated vitamin D3 will be used to try and limit calcium and phosphate absorption while acting on the vitamin D receptor on the parathyroid gland to reduce PTH secretion. Doses may range from 0.25µg two to three times weekly to 4–5µg two to three times weekly.
Side effects include increased calcium and phosphate levels[22],
[23]
, which may require dose reductions or withdrawal of therapy.
Paricalcitol is a newer vitamin D analogue (19-nor-1alpha, 25dihydroxy vitamin D2) and was introduced as a less calcaemic and phosphataemic vitamin D analogue. A small study directly comparing alfacalcidol with paricalcitol in dialysis patients showed a smaller PTH reduction with paricalcitol if the baseline PTH was higher, whereas alfacalcidol suppressed hyperparathyroidism independent of baseline PTH. There was no difference in the incidence of hypercalcaemia or hyperphosphataemia[24]
. Although not formally assessed by NICE, paricalcitol has previously been rejected by the Scottish Medicines Consortium[25]
.
Native vitamin D therapy is recommended by the KDOQI guidelines in patients with stage 3–4 CKD if the PTH is above target range and the vitamin D level is <30ng/mL[26]
. However, there is a lack of evidence to suggest target treatment levels, starting levels, dosing regimes and overall benefits in the CKD population. A systematic review looking at vitamin D use in CKD found significant PTH reduction in stage 3–4 CKD but treatment regimes varied[27]
. Doses of cholecalciferol or ergocalciferol (not available as an oral preparation in the UK) were usually weekly for initiation, and then reduced to monthly. It would seem reasonable to load with weekly dosing, as peak serum levels are seen at seven days and any hypercalcaemia can be detected early and therapy stopped. Native vitamin D has a prolonged half-life of 50 days and so monitoring of calcium and phosphate is essential in CKD patients. There is still much debate around the use of native vitamin D in stages 5–5D CKD, the discussion of which is beyond the scope of this article.
For patients who have refractory secondary hyperparathyroidism with high or normal calcium and a high PTH, the options are either parathyroidectomy or cinacalcet therapy.
Cinacalcet is an agonist of the calcium-sensing receptor on the parathyroid gland, increasing the sensitivity of the gland to extracellular calcium; it is therefore known as a calcimimetic. Because of this action, a significant drop in calcium may be seen when treatment is initiated and calcium levels must be checked within one week of therapy initiation, and after any dose amendments. The majority of side effects seen with cinacalcet are gastrointestinal and those related to hypocalcaemia, such as paraesthesias and muscle cramps. PTH must be monitored every two to four weeks and therapy titrated as appropriate. PTH must be measured at least 12 hours after a dose of cinacalcet because of the initial drop in PTH, which then rises again to a peak at 12 hours[28]
. Patients should be counselled to take their cinacalcet with food as it increases bioavailability and also reduces nausea and vomiting. Cinacalcet can be poorly tolerated because of gastrointestinal effects. Currently there is an intravenous calcimimetic, etelcalcetide, under review by the US Food and Drug Administration (FDA) that is designed to be administered three times weekly after haemodialysis to reduce gastrointestinal side effects.
Cinacalcet is only licensed for use in dialysis patients but in certain circumstances it may be prescribed to stage 4 CKD and post-transplant patients. Hyperparathyroidism may resolve or improve in the following 12 months after renal transplant. However, if parathyroid gland hyperplasia is severe, no improvement may be seen and surgery would then be indicated.
NICE recommends the use of cinacalcet in dialysis patients only if parathyroidectomy is contraindicated and patients have a PTH >85pmol/L with a normal or high-adjusted calcium[29]
, although its use in the dialysis population may be more widespread.
Parathyroidectomy would be indicated in patients with: calciphylaxis, pathological fractures, severe calcifications or hypercalcaemia, hyperphosphataemia and PTH >85pmol/L[30]
. The surgical approaches include subtotal parathyroidectomy (removal of three and a half glands leaving half in situ), total parathyroidectomy with autotransplantation, or total parathyroidectomy without autotransplantation. Autotransplantation involves implantation of some of the healthy parathyroid gland tissue into a different site, usually the non-dominant forearm[30]
. The aim of keeping some active parathyroid tissue is to prevent hypoparathyroidism. The main complication of surgery is hypocalcaemia, which occurs in up to 20% of patients, and so they should be started on calcium supplements and active vitamin D after surgery. ‘Hungry bone syndrome’ can occur immediately post-operatively because of a rapid drop in PTH levels with accelerated bone mineralisation. This is a short-lived occurrence and so supplements and calcium levels need to be reviewed prior to discharge and in the next few days.
Treatment of osteoporosis
Bisphosphonates are not licensed for patients with CrCl <30ml/min. Evidence for their use in osteoporosis in patients with stage 4–5 CKD is limited. One study looked at 301 patients taking risedronate 5mg daily for two years in patients with stage 4 CKD and found no increase in adverse effects[31]
. The main concern with prolonged bisphosphonate use is accumulation leading to adynamic bone disease, and there have been reports to this effect[32]
. Adynamic bone disease should be excluded prior to initiation of therapy in stage 4–5 CKD. Some UK centres use half doses of bisphosphonates (e.g. alendronic acid 70mg every two weeks) in patients with severe CKD[33]
.
Contraindications to therapy
In patients with hypoparathyroidism, active vitamin D, native vitamin D and calcium supplements/binders should be avoided. The calcium may further reduce PTH secretion and increase the risk of adynamic bone disease.
Hypercalcaemic patients should have their calcium binders or supplements stopped and alternative non-calcium-containing binders should be considered. Native vitamin D should be stopped and active vitamin D may need to be reduced, or stopped, depending on the serum calcium level.
In summary, CKD-MBD is a highly complex process caused by derangements in calcium, phosphate, PTH, active vitamin D and FGF-23. To prevent the complications of CKD-MBD, such as fractures and cardiovascular complications, measures to target these derangements are used. These measures include dialysis, dietary restrictions, phosphate binders, active vitamin D3 and, occasionally, calcimimetics or parathyroid gland surgery. Compliance with therapies can be a problem because of palatability and adverse effects; these should be checked with patients and alternatives recommended as appropriate.
Kathrine Parker is senior renal pharmacist and Jane Alderdice is lead renal dietitian, Central Manchester Foundation Hospitals Trust.
Financial and conflicts of interest disclosure: the authors have no relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. No writing assistance was utilised in the production of this manuscript.
Reading this article counts towards your CPD
You can use the following forms to record your learning and action points from this article from Pharmaceutical Journal Publications.
Your CPD module results are stored against your account here at The Pharmaceutical Journal. You must be registered and logged into the site to do this. To review your module results, go to the ‘My Account’ tab and then ‘My CPD’.
Any training, learning or development activities that you undertake for CPD can also be recorded as evidence as part of your RPS Faculty practice-based portfolio when preparing for Faculty membership. To start your RPS Faculty journey today, access the portfolio and tools at www.rpharms.com/Faculty
If your learning was planned in advance, please click:
If your learning was spontaneous, please click:
References
[1] KDIGO Clinical Practice Guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease – Mineral and Bone Disorder (CKD-MBD). Kidney Int 2009;76:S113. Available at: http://www.kdigo.org/pdf/KDIGO%20CKD-MBD%20GL%20KI%20Suppl%20113.pdf (accessed December 2015).
[2] Moe S, Drüeke T, Cunningham J et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease Improving Global Outcomes (KDIGO). Kidney Int 2006;69(11):1945–1953. doi: 10.1038/sj.ki.5000414
[3] Elder G. Pathophysiology and recent advances in the management of renal osteodystrophy. Journal of Bone and Mineral Research 2002;17(12):2094–2105. doi: 10.1359/jbmr.2002.17.12.2094
[4] Martin KJ & Gonzalez EA. Pathophysiology of renal osteodystrophy. Clinical Reviews in Bone and Mineral Metabolism 2007;5(1):11–19. doi: 10.1007/bf02736667
[5] Martin KJ & Gonzalez EA. Metabolic bone disease in chronic kidney disease. Journal of the American Society of Nephrology 2007;18(3):875–885. doi: 10.1681/asn.2006070771
[6] Tanaka Y & Deluca HF. The control of 25-hydroxyvitamin D metabolism by inorganic phosphorus. Arch Biochem Biophys 1973;154(2):566–574. doi: 10.1016/0003-9861(73)90010-6
[7] Goodman WG. Medical management of secondary hyperparathyroidism in chronic renal failure. Nephrol Dial Transplant 2003;18(3):iii2–8. doi: 10.1093/ndt/gfg1002
[8] Gogusev J, Duchambon P, Hory B et al. Depressed expression of calcium receptor in parathyroid gland tissue of patients with hyperparathyroidism. Kidney Int 1997;51(1):328–336. doi: 10.1038/ki.1997.41
[9] Steddon S & Sharples E. Clinical practise guideline CKD-MBD. Renal Association guidelines 2015. Available at: www.renal.org (accessed December 2015).
[10] Stubbs J, Liu S & Quarles LD. Role of fibroblast growth factor 23 in phosphate homeostasis and pathogenesis of disordered mineral metabolism in chronic kidney disease. Semin Dial 2007;20(4):302–308. doi: 10.1111/j.1525-139x.2007.00308.x
[11] Quarles LD. Role of FGF23 in vitamin D and phosphate metabolism: implications in chronic kidney disease. Exp Cell Res 2012;318(9):1040–1048. doi: 10.1016/j.yexcr.2012.02.027
[12] Gutiérrez OM, Mannatadt M, Isakova T et al. Fibroblast growth factor 23 and mortality among patients undergoing haemodialysis. N Engl J Med 2008;359(6):584–592. doi: 10.1056/nejmoa0706130
[13] Kendrick J1, Cheung AK, Kaufman JS et al; HOST Investigators. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J Am Soc Nephrol 2011;22(10):1913–1922. doi: 10.1681/asn.2010121224
[14] NICE clinical guideline 157. Management of hyperphosphataemia in patients with stage 4 or 5 chronic kidney disease 2013. Available at: https://www.nice.org.uk/guidance/cg157 (accessed December 2015).
[15] Jamal S, Vandermeer B, Raggi P et al. Effect of calcium-based versus non-calcium based phosphate binders on mortality in patients with chronic kidney disease: an updated systematic review and meta-analysis. The Lancet 2013;382(9900):1268–1277. doi: 10.1016/s0140-6736(13)60897-1
[16] Daugirdas JT, Finn WF, Emmett M et al. The phosphate binder equivalent dose. Semin Dial 2011;24(1):41–49. doi: 10.1111/j.1525-139x.2011.00849.x
[17] Salusky IB, Foley J, Nelson P et al. Aluminum accumulation during treatment with aluminum hydroxide and dialysis in children and young adults with chronic renal disease. N Engl J Med 1991;24(8): 527–531. doi: 10.1056/nejm199102213240804
[18] Manufacturers Summary of Product Characteristics for Sevelamer carbonate. Consilient Health Ltd. Available at: www.medicines.org.uk (accessed December 2015).
[19] Manufacturers Summary of Product Characteristics for Lanthanum Carbonate. Shire Pharmaceuticals. Available at: www.medicines.org.uk (accessed December 2015).
[20] Manufacturers Summary of Product Characteristics for calcium acetate. Pharmacosmos UK. Available at: www.medicines.org.uk (accessed December 2015).
[21] Musci I, Hercz, G, Uldall R et al. Control of phosphate without any phosphate binders in patients treated with nocturnal haemodialysis. Kidney Int 1998;53(5):1399–1404. doi: 10.1046/j.1523-1755.1998.00875.x
[22] Palmer SC, McGregor DO, Craig JC et al. Vitamin D compounds for people with chronic kidney disease requiring dialysis. Cochrane Database of Systematic Reviews 2009:4. Available at: http://www.cochrane.org/CD005633/RENAL_vitamin-d-compounds-for-people-with-chronic-kidney-disease-requiring-dialysis (accessed December 2015).
[23] Palmer SC, McGregor DO, Craig JC et al. Vitamin D compounds for people with chronic kidney disease not requiring dialysis. Cochrane Database of Systematic Reviews 2009:4. Available at: http://www.cochrane.org/CD008175/RENAL_vitamin-d-compounds-for-people-with-chronic-kidney-disease-not-requiring-dialysis (accessed December 2015).
[24] Hansen D, Rasmussen K, Danielson H et al. No difference between alfacalcidol and paricalcitol in the treatment of secondary hyperparathyroidism in haemodialysis patients: a randomized crossover trial. Kidney Int 2011;80(8):841–850. doi: 10.1038/ki.2011.226
[25] Scottish Medicines Consortium review of paricalcitrol 2008, 478/08. Available at: https://www.scottishmedicines.org.uk/files/paricalcitol_capsules__Zemplar___FINAL_JUNE_2008.doc_for_website.pdf (accessed December 2015).
[26] Kramer H, Berns J, Choi M et al. 25-Hydroxyvitamin D Testing and Supplementation in CKD: An NKF-KDOQI Controversies Report. Am J Kidney Dis 2014;64(4):499–509. doi: 10.1053/j.ajkd.2014.05.018
[27] Kandula P, Dobre M, Schold JD et al. Vitamin D supplementation in Chronic Kidney Disease: a systematic review and meta-analysis of observational studies and randomized controlled trials. Clinical Journal of the American Society of Nephrology 2010;6(1):50–62. doi: 10.2215/cjn.03940510
[28] Wu-Wong JR, Nakane M, Chen Y et al. Mechanistic analysis for time-dependent effects of cinacalcet on serum calcium, phosphorus, and parathyroid hormone levels in 5/6 nephrectomized rats. Physiological Reports 2013;1(3):e00046. doi: 10.1002/phy2.46
[29] NICE technology appraisal guidance [TA117]. Cinacalcet for the treatment of secondary hyperparathyroidism in patients with end-stage renal disease on maintenance dialysis therapy 2007. Available at: https://www.nice.org.uk/guidance/ta117/chapter/1-guidance (accessed December 2015).
[30] Pitt SC, Sippel RS & Chen H. Secondary and tertiary hyperparathyroidism, state of the art surgical management. Surgical Clinics of North America 2009;89(5):1227–1239. doi: 10.1016/j.suc.2009.06.011
[31] Miller PD, Roux C, Boonen S et al. Safety and efficacy of risedronate in patients with age-related reduced renal function as estimated by the Cockcroft and Gault method: a pooled analysis of nine clinical trials. Journal of Bone and Mineral Research 2005;20(12):2105–2115. doi: 10.1359/jbmr.050817
[32] Amerling R, Harbord NB, Pullman J et al. Bisphosphonate use in chronic kidney disease: association with adynamic bone disease in a bone histology series. Blood Purif 2010;29(3):293–299. doi: 10.1159/000276666
[33] UKMI Q&A 169.4. Can oral bisphosphonates be given to people with renal impairment to treat osteoporosis? 2012. Available at: www.medicinesresources.nhs.uk/upload/documents/Evidence/Medicines%20Q%20&%20A/bisphosphonates_q_and_a_final_august_21_2012[1].doc (accessed December 2015).

