

This content was published in 2001. We do not recommend that you take any clinical decisions based on this information without first ensuring you have checked the latest guidance.
The rapid changes occurring in the treatment of human immunodeficiency virus (HIV) infection mean that articles and guidelines on the subject soon become obsolete.
This article provides an overview of current practice and is divided into two parts:
- The use of antiretroviral drugs to control viral replication
- The treatment of HIV-associated opportunistic infections
Antiretroviral therapy

NRTIs
PIs
NNRTIs
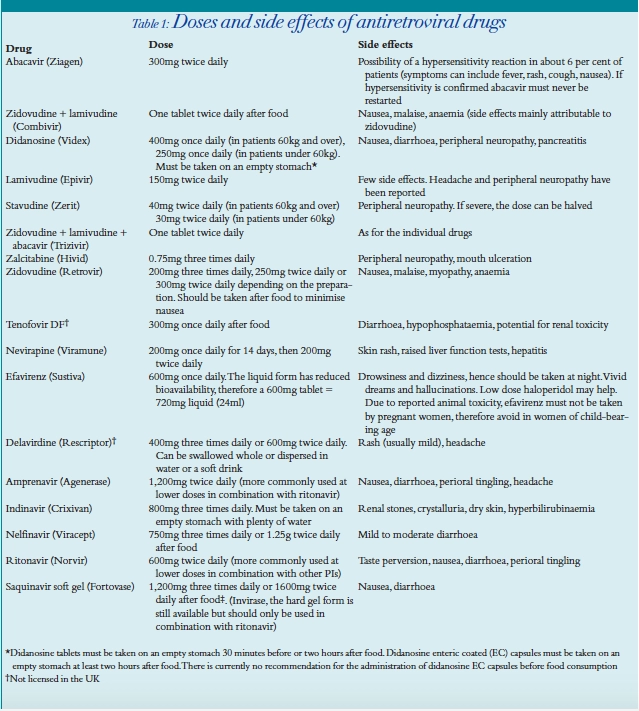
BHIVA guidelines
When to start therapy
- HIV disease state
- Surrogate markers (that is, CD4 count and viral load)
- Assessment of compliance and risk of drug toxicities

What therapy to use
- Likelihood of adherence
- Initial viral load or CD4
- Probability of toxicity
- Co-morbid conditions, such as hepatitis B infection
Table 3 shows the main types of combination antiretroviral therapy used as initial therapy.

Most data are available from studies of two NRTIs and one PI, which have demonstrated a reduced morbidity and mortality in patients treated with a combination containing these two classes of drugs. However, this combination is associated with significant toxicity, especially dyslipidaemia and lipodystrophy. On the other hand, two NRTIs and one NNRTI are associated with less dyslipidaemia, but not as much long-term survival data are available yet. Furthermore, efavirenz has been associated with foetal abnormalities and thus might not be appropriate in women of child-bearing age. The use of three NRTIs is increasingly in vogue because the patient does not have the burden of taking too may tablets. Trizivir is a combination of abacavir, lamivudine and zidovudine in a single tablet. In addition, use of three NRTIs reserves the NNRTI and PI classes for later use. However, there is some debate as to whether three NRTIs are effective in patients with viral loads greater than 100,000 copies per ml.8
The bioavailability of current PIs can be increased by the concomitant use of ritonavir. This factor can be used in overcoming drug resistance and in treating patients with a high initial viral load. The additive or synergistic antiviral effects of PIs in combination might improve potency, relative to a regimen using a single PI. However, the dual PI combination has the disadvantages of increased risk of lipodystrophy, lipid abnormalities and unfavourable drug interactions.
Virological failure
- Lack of potency
- Poor pharmacokinetics (poor absorption, increased metabolism, poor penetration of CSF and seminal fluid, effect of p-glycoprotein, plasma protein binding, drug interactions)
- Host factors, such as non adherence
HIV resistance
HIV resistance testing
Phenotypic assays
Genotypic assays

Treatment of virological failure
Failure of first regimen

Salvage therapy
Discontinuation of therapy is controversial. In most patients, stopping HAART results in a rapid rise in the viral load to pretreatment levels and a fall in the CD4 count. This option must be considered with caution but is an option if the side effects are pronounced.
Drug interactions
Understanding the potential for drug interactions is very important in patients taking HAART.
Both PIs and NNRTIs are metabolised exclusively in the liver by the cytochrome P450 (CYP) isoenzyme system. The nucleoside analogues are mainly cleared renally and so do not pose as many metabolic drug interaction problems. Identification is now possible in early drug development of the dominant cytochrome P450 isoforms responsible for metabolising a particular drug. In addition, there is the ability to detect whether a drug is capable of inhibiting or inducing a specific CYP isoform.
The major isoforms involved in human drug metabolism are CYP3A, CYP2D6, CYP2C, CYP1A2 and CYP2E1.9 Patients who are HIV-positive often take many drugs concurrently, hence an assessment of potential drug interactions is imperative.
CYP3A is the major isoform responsible for all PI and NNRTI metabolism. Coadministration of enzyme inducers leads to a danger of lowered drug levels of the PIs and NNRTIs, thus increasing the risk of development of drug resistance. Conversely, co-administration of enzyme inhibitors could lead to increased toxicity.
Treatment of tuberculosis (TB) in HIV-positive patients taking HAART poses complications because rifampicin, the main component of any anti- TB regimen, is probably one of the most potent enzyme inducers known. Rifampicin is therefore not to be taken with most of the PIs and NNRTIs, although rifabutin can be used as an alternative in some cases, with careful dose adjustment. It is essential that the product literature is consulted if rifampicin or rifabutin are co-prescribed with anti-HIV drugs.
All the PIs inhibit the isoenzyme CYP3A4,which makes co-administration of drugs such as astemizole and terfenadine contraindicated, owing to the risk of arrhythmias. Some PIs and NNRTIs are also capable of inducing isoenzymes, making coadministration of the combined oral contraceptive pill inadvisable as a sole method of contraception.
The potential for enzyme induction and inhibition with all the PIs and NNRTIs causes complications when constructing a HAART regimen using combinations of both classes of drugs. Referral to the product literature is vital to ensure that any dose modifications required are applied.
Of all the PIs, ritonavir is the most potent inhibitor of the CYP3A isoenzyme. It also inhibits P-glycoprotein, a transport protein responsible for pumping out certain drugs from the gut lining, thereby preventing their absorption. Since the full dose of ritonavir is associated with several side effects, it is now more commonly used in combination with other PIs, mainly for its enzyme inhibition properties. It acts to optimise the pharmacokinetic profile of co-administered PIs, either by reducing first pass metabolism, or by increasing their half lives. Co-administration of low dose ritonavir can therefore often simplify the HAART regimen.For example, the conventional dose of indinavir is 800mg eight-hourly, but by adding low dose ritonavir twice daily, the indinavir can be reduced to twice daily dosing. The food restrictions associated with indinavir disappear with this regimen and patients are more easily able to comply.
Emerging toxicities
Previously unknown toxicities of antiretroviral drugs are being discovered as more experience is gained in their use. These include mitochondrial toxicity, lactic acidosis and lipodystrophy.
Mitochondrial toxicity
Mitochondrial toxicity is a relatively new term used to describe a host of disorders thought to be attributable to nucleoside analogues. NRTIs are thought to inhibit the human mitochondrial enzyme, deoxyribonucleic acid (DNA) polymerase gamma in a similar way to their action against HIV, resulting in the production of dysfunctional mitochondria.10 Pancreatitis and peripheral neuropathy (which are well known side effects of some of the NRTIs) are now thought potentially to result from mitochondrial toxicity. In addition, the Committee on Safety of Medicines issued a summary in June, 1999, of eight cases of mitochondrial dysfunction in infants exposed antenatally to zidovudine with or without lamivudine. However, the committee concluded that there were insufficient data to establish a causal relationship between nucleoside analogue exposure and mitochondrial dysfunction.11
Lactic acidosis
Lactic acidosis is also emerging as a new toxicity in patients taking HAART. It is postulated that antiretroviralassociated lactic acidosis is also a manifestation of mitochondrial toxicity arising from the use of nucleoside analogues. Lactic acidosis is a known toxicity of NRTIs, thus the summaries of product characteristics (SPCs) of all the nucleoside analogues warn of the risk of lactic acidosis. Although no causal relationship has been formally linked to any of the NRTIs in particular, it is postulated that the incidence is more likely with the use of stavudine and didanosine.
Although patients can be asymptomatic, prominent symptoms include weight loss, fatigue, nausea, bloating, vomiting and abdominal pain.
Associated examination and laboratory abnormalities can include:
- Tachycardia and hypotension
- Abnormal liver function tests (both transaminases and alkaline phosphatase)
- Low chloride, low bicarbonate, raised anion gap
- Raised glucose
- Raised creatine kinase
- Raised amylase
The European Medicines Evaluation Agency has been made aware of seven cases of lactic acidosis in women treated during pregnancy with the combination of stavudine and didanosine. At present, there is insufficient information to decide whether pregnancy is an additional risk factor for lactic acidosis. It is also uncertain whether any increased risk of lactic acidosis is specific to stavudine and didanosine or whether it might be increased with all combinations of NRTIs.12 However, the use of these two drugs during pregnancy should be avoided, and indeed, where possible, zidovudine should be used as a component of the NRTI portion of the regimen to prevent HIV vertical transmission.
Lipodystrophy
One of the main drawbacks to protease inhibitor therapy is the risk of lipodystrophy. This syndrome is now well described with PIs and encompasses a range of manifestations, such as body fat redistribution, hyperlipidaemia, insulin resistance and diabetes mellitus. Many patients are reluctant to take PIs because of the changes in body shape that they can induce. These changes sometimes make it obvious that a patient is on anti-HIV therapy.
The characteristic signs are loss of fat from the face and limbs, accumulation of visceral fat around the abdomen and occasional formation of a “buffalo hump”. It is postulated that this syndrome is caused by PIs somehow interfering with the process of fat metabolism, as a result of their effect on the cytochrome P450 enzyme system in the liver. However, more than one theory has been put forward and the exact mechanism is still unknown.
It has been suggested that inhibition of mitochondrial DNA synthesis (hence mitochondrial toxicity) might play some role in the development of lipodystrophy, following case reports in patients not taking PIs, but taking NRTIs.13 Lipoatrophy (loss of fat from the face and limbs) has been linked, anecdotally, to the use of NRTIs, especially stavudine, although there is no strong evidence to support this at present. To most patients, the fat redistribution syndrome on the whole is still strongly thought of as being related to protease inhibitor therapy.
Abnormalities of lipid metabolism in patients infected with HIV were described prior to the advent of HAART, including elevated triglycerides and a reduction in the levels of high density lipoprotein (HDL) cholesterol.14 Additional significant increases in triglyceride and both low density lipoprotein (LDL) and very low density lipo-protein (VLDL) cholesterol concentrations have been associated with all the available protease inhibitors. However, lipid elevations have also been reported in patients receiving NNRTIs, albeit less frequently. It is not yet known if the lipid elevations that exist during HAART represent a significant risk factor for cardiovascular disease. These observations are a reason for increased concern for the eventual increase in cardiovascular complications as mortality associated with AIDS falls due to HAART.
Treatment of hypercholesterolaemia is usually with 3-hydroxy-3-methylglutaryl co-enzyme A (HMG CoA) reductase inhibitors. However, in HIV-infected patients taking HAART, careful consideration needs to be given to potential drug interactions with the statins. Simvastatin is now contraindicated with protease inhibitors due to the risk of myalgia,myositis and myopathy, caused by an increase in simvastatin levels brought about by enzyme inhibition. Low dose atorvastatin can be used — its metabolism is also inhibited by protease inhibitors, but to a lesser extent. Pravastatin can be used as it is not metabolised by the CYP isoenzyme system, although high doses are often required to achieve adequate response. In the treatment of hypertriglyceridaemia there are no drugdrug interactions with the co-administration of fibrates and HAART.
One published study suggests that patients established on PIs with an undetectable viral load can achieve some resolution of their dyslipidaemia by switching to a nevirapinebased regimen, without losing viral control.15 Other such “switch” studies have been reported in which efavirenz or abacavir were used as the drugs of choice, although efavirenz has failed to demonstrate a consistent benefit.16
New therapies
Lopinavir is the latest addition to the PIs. It is co-formulated with ritonavir (as Kaletra, recently launched in the UK), and the latter acts as a pharmacokinetic enhancer, substantially increasing lopinavir drug exposure. The area under the plasma concentration-time curve (AUC) is increased 100-fold compared with that for lopinavir alone. This provides a pharmacological barrier to the emergence of viral resistance and the degree of drug exposure attained is sufficient to suppress the replication of viral strains that are genotypically or phenotypically resistant to the drug.17 The benefits of elevated drug concentrations have to be weighed against the risks of short or long-term toxicity. At the dose selected for phase III clinical trials (400mg lopinavir and 100mg ritonavir twice daily), it appears to be well tolerated and has been shown, at least in the short-term, not to have many major side effects.
Fusion inhibitors
T20 belongs to an entirely new class of antiretrovirals called fusion inhibitors and is derived from a protein called gp41, the HIV protein which penetrates uninfected cells as the first step in viral entry. T20 is believed to interfere with this cell entry process. It is administered subcutaneously twice daily as part of a HAART regimen. At present, in the UK, it is only available in clinical trials for patients whose treatment options have been limited by drug resistance.
Tenofovir
Tenofovir, a nucleotide reverse transcriptase inhibitor, has recently been made available on a named-patient basis in the UK. It acts in exactly the same way as NRTIs. However, its structure contains an extra phosphate group, so it does not require triphosphorylation in order to become active within the cell. It has the advantage that it is administered as a single tablet once a day, and is thought to remain active against viruses that have become resistant to many of the NRTIs.
Nucleotide analogues such as tenofovir have the potential to cause renal toxicity, hence co-administration of renally toxic drugs is currently contraindicated with them. Renally toxic drugs, including foscarnet, cidofovir, aminoglycosides, and amphotericin, are used quite commonly in HIV-infected patients and therefore could be a potential problem.
Immunomodulatory agents
Immunomodulatory agents include interleukin-2 and interferon-alpha.
Interleukin-2 (IL-2)
The cytokine IL-2 is administered in cycles of twice-daily subcutaneous injections for five days every eight weeks. IL-2 has been shown to increase the CD4 count with improved lymphocyte responses.
Currently, a large multicentre trial is being conducted to determine whether there is an improvement in morbidity or mortality with the use of IL-2.
There is a possible role for IL-2 in patients who have achieved an undetectable viral load with HAART, but whose CD4 count is still low, thus putting them at risk of opportunistic infections.
Interferon-alpha
Interferon-alpha is a cytokine whose use is being investigated as adjuvant therapy in salvage HAART regimens.
Opportunistic infections
Drug research has tended to concentrate on new antiretrovirals over recent years. Research into new drugs for the treatment of opportunistic infections (OIs) has become less attractive to pharmaceutical companies, with the introduction of HAART seeing a reduction in incidence. There are two main elements to the management of OIs — prevention and treatment.
HIV-infected patients with a CD4 count less than 200 cells per mm3 are at greatest risk of OIs. Table 6 shows the recommended agents for prophylaxis against the common HIV-associated infections.
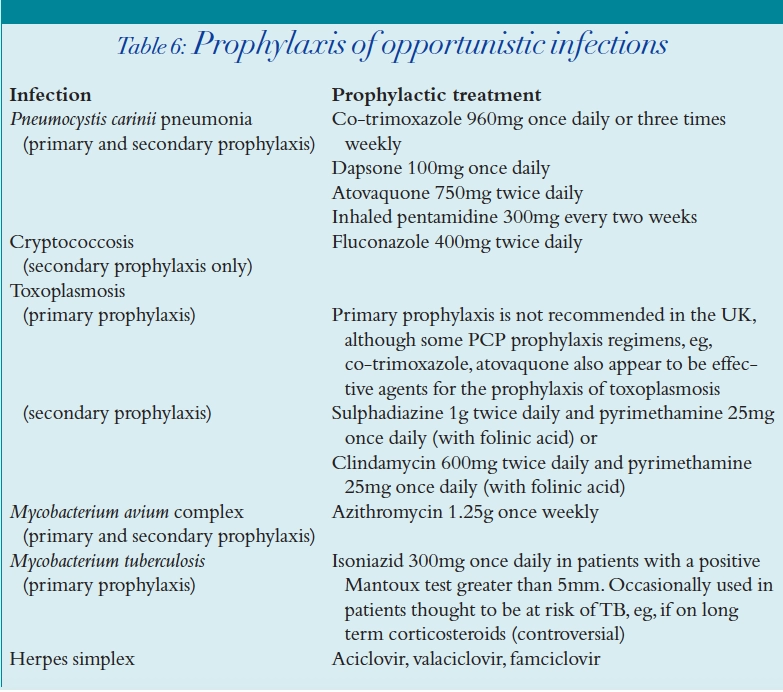
Prophylaxis can be stopped for several of the OIs once the CD4 count has risen in a sustained manner. For example Pneumocystis carinii pneumonia (PCP) prophylaxis is commonly discontinued after the CD4 count has been above 200 cells per mm3 for three to six months.18
The treatments available for different OIs are outlined below.
PCP
Several agents have been successfully used in the treatment of PCP (Table 7).

Co-trimoxazole is the “gold standard” for the treatment of PCP. It should be used first line unless there has been any previous allergy to the drug.
Clindamycin-primaquine should be considered in those patients with co-trimoxazole allergy. This regimen cannot be used in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency.
Atovaquone can be used, but it is not as effective as other regimens in treating severe PCP, thus it should only be used in mild to moderate infection.
Pentamidine tends to be reserved for cases where other treatment has failed or where there are no other options. Patients should have inhaled pentamidine administered in conjunction with IV therapy for the initial three days as it takes time to achieve adequate plasma levels of the drug.
Use of trimetrexate should be considered as salvage therapy when patients have failed to respond to conventional treatment. It is highly toxic to the bone marrow and therefore requires high doses of folinic acid rescue therapy.
Corticosteroids should be used as adjuvant therapy to PCP treatment in patients with PaO2 lower than 8kPa because they accelerate the recovery process and prevent the development of respiratory failure and death.19 A typical regimen is methylprednisolone IV given initially every six hours, which is then converted to an equivalent oral prednisolone dose and reduced slowly over the treatment period.
Toxoplasma gondii encephalitis
Treatment of T. gondii encephalitis involves pyrimethamine plus sulphadiazine or clindamycin. The regimens are:
- Sulphadiazine 2g IV or orally four times a day + pyrimethamine 75mg once daily (+ folinic acid)
- Clindamycin 600mg IV or orally four times a day + pyrimethamine 75mg once daily (+ folinic acid)
Both these regimens exhibit a high incidence of adverse events and involve large quantities of tablets. Atovaquone has activity against toxoplasmosis and can be used if side effects or compliance are an issue. Corticosteroids can be used in patients who have evidence of cerebral oedema and increased intracranial pressure. Initial treatment of toxoplasmosis should be for six weeks, after which continuous maintenance therapy is required (secondary prophylaxis).
Cytomegalovirus (CMV) infection
CMV infections are still significant problems in HIV disease, specially in the more severely immunocompromised patients (with CD4 counts lower than 50 cells per mm3).20 The antiviral agents used to treat CMV infection are ganciclovir, foscarnet or cidofovir, intravenously. All three of these antiviral agents are licensed for the treatment of CMV retinitis, although not all are licensed for the treatment of CMV infection elsewhere in the body. First line therapy is usually intravenous ganciclovir, administered twice daily for a period of 14 to 21 days, depending on the site of infection. In CMV retinitis, a maintenance regimen must then be chosen for the patient, to prevent reactivation of disease and risk of blindness. (This is not normally required for infection in other sites.) The maintenance phase for CMV retinitis is indefinite or until an adequate CD4 count rise has been achieved with HAART.21 Maintenance treatment can often be safely discontinued once the CD4 count has risen above 100 cells per mm3. Systemic maintenance therapy should be used where possible, usually oral ganciclovir. If additional local therapy is required, intravitreal injections of ganciclovir or foscarnet or a ganciclovir ocular implant may be used. Fomivirsen is a novel antiviral drug that is only available as an injection for intravitreal administration. Cidofovir is an antiviral agent that is active against ganciclovir-resistant CMV. It is administered intravenously once a week for two consecutive weeks as a loading dose, then once every fortnight. This is licensed for second line treatment of CMV retinitis. However, it is sometimes used as an alternative maintenance therapy if adherence to oral ganciclovir is an issue. Cidofovir has high renal toxicity, therefore patients must be kept well hydrated with normal saline. Probenecid is administered to delay renal excretion of cidofovir, so preventing renal tubular damage. Patients receiving cidofovir are also at risk of uveitis and hypotony, which means that they need to be monitored regularly. Cidofovir is not administered intravitreally because of the risk of irreversible hypotony.
Mycobacterium avium complex (MAC)
MAC is common in AIDS and is associated with an accelerated rate of disease progression.22 As MAC is prone to develop drug resistance, combination therapy is mandatory. Regimens should consist of a macrolide antibiotic (clarithromycin or azithromycin) with at least one other antimicrobial agent, usually ethambutol. Rifabutin is sometimes used in addition, although less so nowadays due to the risk of drug interactions with the antiretrovirals agents, and a combination of two drugs is usually adequate. Other drugs with activity against MAC include amikacin and ciprofloxacin. Table 8 shows the doses at which these drugs are used.
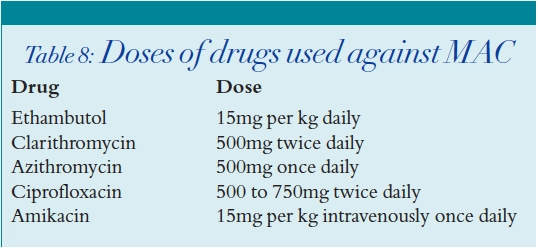
Mycobacterium tuberculosis
The standard antituberculous therapy for HIV-infected individuals with fully sensitive tuberculosis (TB) consists of isoniazid, rifampicin,pyrazinamide and ethambutol for two months, followed by isoniazid and rifampicin for four months.
Treatment of TB in HIV-infected patients is always a complicated issue due to the risk of drug interactions with rifamycins and HAART. Rifampicin is not currently recommended to be used in combination with any of the PIs or NNRTIs, with the exception of efavirenz. There are some pharmacokinetic data to support the use of rifampicin with a raised dose of efavirenz (800mg daily instead of the usual 600mg daily dose).23 Rifabutin is commonly used to treat TB in HIV patients taking HAART as it may be used with some of the PIs and NNRTIs, providing dose adjustments are applied. It is important to refer to the antiretroviral product liter ature before initiating rifamycin therapy.
Drugs used in the treatment of other HIV-related OIs are listed in Panel 2.

Malignancies
Since the introduction of HAART, the incidence of AIDS-related malignancies such as Kaposi’s sarcoma and primary cerebral lymphoma have declined3 while that of non-Hodgkin’s lymphoma has remained unchanged.24, the need for effective treatments for these malignancies remains.
Kaposi’s sarcoma
First line therapy involves starting HAART. HAART alone can achieve complete remission of Kaposi’s sarcoma (KS) lesions in up to 50 per cent of patients.25 In some patients, the KS lesions can worsen initially as part of an immune restoration illness. If after three to four months the KS has not improved, or a more rapid response is needed, for example, in the presence of oedema, visceral disease or extensive disfiguring lesions, then systemic chemotherapy should be considered. The first line treatment is liposomal anthracyclines and salvage therapy is with paclitaxel. An alternative is interferon-alpha if the CD4 count is above 400 cells per mm3.
Primary cerebral lymphoma
Primary cerebral lymphoma is associated with advanced immunosuppression. It can be difficult to diagnose and the patient has a median survival of only two to four months. There are no curative therapies and so many patients opt for symptom palliation alone. Standard therapy is whole brain irradiation, although one cohort study supports the use of high dose intravenous methotrexate in patients with good performance status.26
Non-Hodgkin’s lymphoma
Early treatment (pre-HAART) of non-Hodgkin’s lymphoma was badly tolerated and had poor results. The use of haemopoietic growth factors, intrathecal chemotherapy prophylaxis, prophylaxis of OIs and HAART have led to improved outcomes. Many centres stratify chemotherapy for patients with preserved immune systems. Regimens used include mBACOD (bleomycin, doxorubicin, vincristine, dexamethasone methotrexate and folinic acid), BEMOP-CA (bleomycin, etoposide, methotrexate, vincristine prednisolone and folinic acid with cyclophosphamide and doxorubicin) and CHOP (cyclophosphamide, doxorubicin, vincristine and prednisolone), although recent experience with cyclophosphamide , doxorubicin, and etoposide infusions (CDE) administered continuously over 96 hours has been promising.27 Drug interactions between chemotherapy and HAART should be considered and doses modified as appropriate. Intrathecal chemoprophylaxis and OI prophylaxis are vital parts of therapy; the management of HIV patients infected with non-Hodgkin’s lymphoma should only be undertaken by a team of clinicians experienced in both medical oncology and HIV clinical care.
Summary
The treatment of HIV disease remains complex and is rapidly changing, requiring the input of specialist heath care professionals. New antiretroviral drugs are being continually developed and the use of currently available drugs are being refined. There have been fewer drug developments in the treatment of OIs over the last few years but these still remain an important aspect of HIV treatment.
References
- Pozniak A. Surrogacy in HIV-1 clinical trials. Lancet 1998; 351:536–7.
- Paterson D, Swindells S, Mohr J, Brester M,Vergis E, Squier C, et al. Adherence to protease inhibitor therapy and outcomes in patients with HIV infection. Ann Intern Med 2000;133:21–30.
- Palella FJ, Delaney KM,Moorman AC, Loveless MO, Fuhrer J, Satten GA, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N Engl J Med 1998;338:853–60.
- Staszewski S, Morales-Ramirez J,Tashima KT, Rachlis A, Skiest D, Stanford J, et al (for the study 006 team). Efavirenz plus zidovudine and lamivudine, efavirenz plus indinavir, and indinavir plus zidovudine and lamivudine in the treatment of HIV- 1 infection in adults. N Engl J Med 1999;341:1865–73.
- Squires K. The Atlantic Study: a randomised, open-label trial comparing two protease inhibitor (PI)-sparing antiretroviral strategies versus a standard PI-containing regimen, 48-week data [abstract]. Proceedings of the Thirteenth International AIDS conference on antimicrobial agents and chemotherapy; 2000 Jul 9–14; Durban. Abstract no L6PeB7046.
- British HIV Association. Guidelines for the treatment of HIV-infected adults with antiretroviral therapy. HIV Medicine. 2000;1:76–100.
- Pontesilli O,Kerkhof-Garde S, Notermans DW, Foudraine NA, Roos M, Klein MR, et al. Functional T-cell reconstitution and human immunodeficiency virus-1-specific cellmediated immunity during highly active antiretroviral therapy. J Infect Dis 1999;180:76–86.
- Staszewski S,Keiser P, Montaner J, Raffi F, Gathe J, Brotas V, et al . Abacavirlamivudine- zidovudine vs indinavirlamivudine- zidovudine in antiretroviral-naive HIV-infected adults: a randomised equivalence trial. JAMA; 2001 285:1155–63.
- Bertz R, Granneman G. Use of in vitro and in vivo data to estimate the likelihood of metabolic pharmacokinetic interactions. Clin Pharmacokinet 1997; 32: 210–58.
- Brinkman K, Smeitink JA, Romijn JA, Reiss P. Mitochondrial toxicity induced by nucleoside-analogue reversetranscriptase inhibitors is a key factor in the pathogenesis of antiretroviral therapyrelated lipodystrophy. Lancet 1999;354:1112–5.
- Breckenridge A. Antiretroviral drugs to reduce vertical transmission of HIV infection.CSM/CMO/99/5.June 25, 1999.
- EMEA public statement. Reports of lactic acidosis in pregnant women treated with Zerit and Videx. January 26, 2001.
- Madge S, Kinloch-de-Loes S,Mercey D, Johnson MA,Weller IV. Lipodystrophy in patients naive to HIV protease inhibitors. AIDS 1999;13:735–7.
- Grunfield,Pang M, Doerrler, Shigenaga JK, Jensen P, Fenigold KR. Lipids, lipoproteins, triglyceride clearance and cytokines in HIV infection and AIDS. J Clin Endocrinol Metab 1992;74:1045–52.
- Ruiz L, Bonjoch A,Paredes R, Johnson S, Arno A, Roneu J, et al. A multi-centre, randomised, open-label comparative trial of the clinical benefit of switching the protease inhibitor by nevirapine in HAART-experienced patients suffering lipodystrophy [abstract]. Proceedings of the Sixth Conference on retroviruses and opportunistic infections; 1999 Feb; Chicago. Abstract no LB-14.
- Moyle G, Baldwin C. Switching From a PI-based to a PI-sparing regimen for management of metabolic or clinical fat redistribution. AIDS Reader 2000;10:479–85.
- Bertz R, Lam W, Brun S,Kumar G, Fields C,Orth K, et al. Multiple-dose pharmacokinetics (PK) of ABT- 378/ritonavir (ABT-378/r) in HIV positive subjects [abstract]. Proceedings of the Thirty-ninth Interscience conference on antimicrobial agents and chemotherapy; 1999 Sept; San Francisco. Abstract no 0327.
- Furrer H, Egger M, Opravil M, Bernasconi E, Hirchel B, Battegay M, et al. Discontinuation of primary prophylaxis against Pneumocystis carinii pneumonia in HIV-1-infected adults treated with combination antiretroviral therapy.N Engl J Med 1999;340:1301–6.
- Montaner J, Lawson L, Levitt N, Belzberg A, Schechter MT, Ruedy J. Corticosteroids prevent early deterioration in patients with moderately severe Pneumocystis carinii pneumonia and the acquired immunodeficiency syndrome. Ann Intern Med 1990;113:14–20.
- Kuppermann BD,Petty JG, Richman DD, Matthews WC, Fullerton SC, Rickman LS et al. Correlation between CD4 counts and prevalence of cytomegalovirus retinitis and human immunodeficiency virus-related noninfectious retinal vasculopathy in patients with acquired immunodeficiency syndrome. Am J Ophthalmol 1993;115:575–82.
- Studies of the ocular complications of AIDS Research Group, in collaboration with the AIDS Clinical Trials Group. Foscarnet-ganciclovir cytomegalovirus retinitis trial 4: visual outcomes. Ophthalmology 1994;101:1250–61.
- Chaisson RE,Moore RD,Richman DD, Kerly J, Creagh T. Incidence and natural history of Mycobacterium aviumcomplex infections in patients with advanced human immunodeficiency virus disease treated with zidovudine. Am Rev Respir Dis 1992;146:285–9.
- DuPont Pharma. Efavirenz summary of product characteristics. Stevenage: DuPont; 1999
- Matthews G,Bower M, Mandalia S, Powles T, Nelson M,Gazzard B. Changes in acquired immunodeficiency syndrome-related lymphoma since the introduction of highly active antiretroviral therapy. Blood 2000;96:2730–4.
- Martinelli C, Zazzi M,Ambu DS, Bartolozzi D, Corsi P, Leoncini F. Complete regression of AIDS-related Kaposi’s sarcoma associated human herpes virus-8 during therapy with indinavir. AIDS 1998;12:1717–9.
- Jacomet C, Girard PM, Lebrette MG, Farese VL, Monfort L, Rozenbaum W. Intravenous methotrexate for primary central nervous system non-Hodgkin’s lymphoma in AIDS. AIDS 1997;11:1725–30.
- Sparano JA,Wiernik PH, Hu X, Sarta C, Schwartz EL, Soeiro R, et al. Pilot trial of infusional cyclophosphamide, doxorubicin, and etoposide plus didanosine and filgrastim in patients with HIV-associated non-Hodgkin’s lymphoma. J Clin Oncol 1996;14:3026–35.
You might also be interested in…

EMA may ask manufacturers to increase production capacity in response to medicines shortages

Preparing for the GPhC registration exams: how to revise learning outcomes relating to clinical knowledge and therapeutic approaches
