The term “dementia” is used to describe a number of diseases, the most common of which is Alzheimer’s disease (AD). Other types of dementia include vascular dementia, dementia with Lewy bodies and frontotemporal dementia, as well as dementias resulting from infections, such as Creutzfeldt-Jakob disease or HIV, and from genetically inherited syndromes, such as Huntington’s disease (Huntington’s chorea).
Dementia is defined by the World Health Organization as “a syndrome due to diseases of the brain, usually of a chronic or progressivenature, in which there are disturbances of multiple higher cortical functions, including memory, thinking, orientation, comprehension, calculation, learning capacity, language and judgement. Consciousness is not clouded. The impairments of cognitive function are commonly accompanied, and occasionally preceded, by deterioration in emotional control, social behaviour or motivation.”[1] However, there is some variation in this definition in practice. For example, patients with dementia with Lewy bodies can experience variations in consciousness (see below). In addition, it is accepted that early stage dementia may be mild and, therefore, not fulfil the WHO criteria.
Some dementias, such as those caused by drugs, alcohol or non-organic psychiatric disorders, are reversible. Most dementias, however, are progressive and incurable. Although, in some cases, medicines can be prescribed to alleviate symptoms, most patients with dementia can expect, eventually, to become unable to communicate and to need help performing “activities of daily living”. Eleven of these activities are essential for self-care, including eating, dressing, walking and looking after personal hygiene.
The main risk factor for dementia is age. Prevalence is 2 per cent in those aged 65–69 years compared with 20 per cent in those aged 85–89.[2] The terms senile and pre-senile dementia have been used to differentiate between patients under or over 65 years but these are no longer in common use because the two types share some aetiological features.
Dementia is estimated to affect 24.3 million people worldwide. There are 4.6 million new cases every year and it is suggested that this figure will double every 20 years, reaching over 80 million by 2040. Although 60 per cent of patients with dementia live in developed countries the rate of increase in developing countries is three to four times that in developed regions, possibly because diagnosis in these countries has improved.[3]
Patients with cognitive impairment consume substantial health resources. For example, in the UK, around 224,000 of these people are in institutional care at an estimated cost of 4.6bn each year.
Diagnosis
The symptoms developed by a patient with dementia depend on the type of dementia and vary with the brain area affected. For example, in frontotemporal dementia behaviour and mood are typically affected. Diagnosis must be made by a specialist and involves a combination of taking a medical history, observation and testing intellectual function and memory. Several assessment scales can be used, the most common being the mini mental state examination (MMSE) (see Box 1).
It is important that the type of dementia is diagnosed correctly because this can affect treatment options. For example, of the four drugs currently available for dementia, three are licensed for dementia in Alzheimer’s disease only.
Box 1: Mini mental state examination
The MMSE consists of a series of tests. It is split into five sections:
- Orientation (eg, the patient is asked “what day of the week is it?”)
- Memory part 1 (eg, the patient is asked to remember three objects)
- Attention and calculation (eg, the patient is asked to subtract five from 50 in a sequence or spell a word backwards)
- Memory part 2 (eg, the patient is asked to recall objects from the first memory test)
- Language, writing and drawing (eg, the patient is asked to follow a set of instructions)
The patient scores points for completing the five sections successfully. MMSE scores are related to age and level of education (eg, the median MMSE score for individuals with at least nine years of schooling is 29) so scores should be adjusted to take these factors into account. The MMSE scale has a maximum of 30 points. Mild dementia is usually associated with an MMSE score of 21–26, moderate dementia with a score of 10–20, and severe dementia with a score less than 10.
Case
Mrs SD is a 72-year-old ex-nurse who was treated for breast cancer 10 years ago. She attended her GP surgery with her husband, which was unusual, complaining of insomnia and lack of interest in family, friends, eating, etc. On examination, Mrs SD appeared mildly depressed. On performing the MMSE she scored 25. She lost 2 points with short term recall, 2 points in spelling “toast” backwards and 1 point with orientation, confusing summer for winter. Her education history suggests that her MMSE score should be over 28 and, in this case, a diagnosis of mild dementia was made.
Alzheimer’s disease
Alzheimer’s disease (AD) accounts for around 60 per cent of all cases of dementia. It is characterised by memory loss, typically progressing to loss of cognition.At a microscopic level, abnormal structures called neurofibrillary tangles and amyloid plaques are observed in brain tissue. These are accompanied by chronic inflammatory processes, neurovascular dysfunction and abnormalities of cell signalling, resulting in degeneration of neurones and loss of synapses — the brains of AD sufferers are usually shrunken due to atrophy.
Neurofibrillary tangles
In normal nerve cells the neuronal cytoskeleton is supported by microtubules which maintain the shape of the axon (see Figure 1) and allow correct transport of nutrients. One of the constituents of this structure is a protein called tau, which binds with the microtubules to form a structurally sound cytoskeleton.
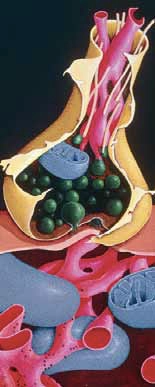
Control of this binding is achieved via enzymatic phosphorylation of binding sites. The more phosphorylation the less association between the tau protein and the microtubule. In AD, tau proteins are hyperphosphorylated resulting in poor microtubule assembly and loss of function. This initially affects neuronal communication, but eventually leads to cell death.
Hyperphosphorylated tau proteins have a tendency to sequester normal tau and other microtubule-associated proteins to form insoluble fibrils, which form tangles. These further impair neuronal function and induce a chronic inflammatory response.
Neuronal damage can begin many years before the symptoms of AD develop. It usually starts in the hippocampus, progressing to the cerebral cortex, and this reflects the typical progression of AD from initial loss of short-term memory to impaired language skills and loss of the ability to take informed, reasoned decisions.
Amyloid plaques
Amyloid plaques are large insoluble structures that develop in extraneuronal spaces. In the 1980s, it was discovered that the cores of these plaques are composed of neurotoxic beta-amyloid protein (βΑ).This protein, initially thought to be an abnormal product, is in fact a product of normal cell metabolism. βΑ is produced in the metabolism of a large transmembranous protein (known as amyloid precursor protein; APP), which can follow two routes: via α-secretase (resulting in a neuroprotective product) or via β-secretase and γ-secretase which produces βΑ. This in itself is not abnormal but, in many people with dementia, βΑ clearance is reduced and it accumulates. This leads to subtle conformational changes in the protein structure and the formation of soluble and insoluble oligomers, which deposit as diffuse plaques.
The formation of βΑ-containing metabolic products depends on a variety of cellular signals, one of which is related to the muscarinic cholinergic receptor. This reduces βΑ formation and the receptor is, therefore, a potential target for preventing disease progression.[4]
Plaque formation is accompanied by inflammatory responses. Neurones in the brain are supported by glial cells, which are classified as either macroglia (astrocytes and oligodendrocytes) or microglia. Microglia multiply in response to injury and infection. Astrocytes and microglia concentrate around amyloid plaques and cause neuronal death, the former via prostaglandin-mediated pathways and the latter via proinflammatory cytokines, such as interleukin-1b, IL-6, and TNF-alpha. Studies have reported elevated TNF-alpha levels in the cerebrospinal fluid and serum of patients with AD. All these pathways are, therefore, possible pharmacological treatment targets.
The part played by APP and the inflammatory cascade in development of dementia is supported by genetic studies that identify patients with familial early onset dementia as having mutations in the genes for both APP and the enzymes responsible for βΑ production. Patients under 65 years old with dementia and amyloid plaques, such as those with Down’s syndrome, express an extra APP gene. Over expression of the APP protein appears to be closely related to the development of familial dementia and causes the overproduction of βΑ.
The classic biochemical theory associated with AD is the cholinergic hypothesis, which states that the disease involves a cholinergic deficit — autopsy data demonstrate that over three-quarters of cholinergic neurons in the basal forebrain nuclei are destroyed in typical AD sufferers.
These microscopic and biochemical features are likely to be closely related and might be interdependent. It has been suggested that altered cerebrovascular microcirculation might cause the reduced clearance of βΑ and the subsequent development of plaques. In turn, the development of plaques causes altered neuronal function as a result of chronic inflammation and subsequent signalling abnormalities, which may be responsible for the excess phosphorylation of tau proteins, leading to neurofibrillary tangles.
A CPD article, to be published on 25 November, will focus on Alzheimer’s disease and its treatment.
Vascular dementia
Vascular dementia (VaD) accounts for up to 20 per cent of all cases of dementia. It is more common in men than in women and is influenced by age. The term covers dementia caused by ischaemic or haemorrhagic cerebrovascular lesions, although ischaemic lesions are the most commonly encountered.[4] Multi-infarct dementia (lesions caused by a series of small strokes) is considered the main type of VaD.
The pathogenic processes at play in the development of VaD are many, affecting numerous cerebral vessels and inducing inflammatory processes. In general, the incidence of VaD doubles every five years. Many risk factors are associated with the condition but the significance of each is uncertain. These include:
- Atrial fibrillation
- Hypercholesterolaemia
- Hypertension
- Smoking
- Diabetes
- Hyperhomocysteinaemia (see Box 2)
- VaD can also have a genetic component
Subtypes of VaD are: large-vessel vascular dementia, small-vessel vascular dementia, haemorrhagic vascular dementia and hypoperfusion vascular dementia. Multi-infarct dementia falls within the large vessel subtype.
VaD tends to have a more sudden onset than AD. Patients can present with acutely developing dementia as a result of a single cerebrovascular event or with dementia progressing throughout the course of a cerebrovascular lesion. The subtype will largely govern symptoms. Large cortical lesions present in a similar way to AD and patients will experience anxiety and depression. Subcortical ischaemic dementia, a type of small-vessel VaD, typically presents with psychomotor slowness often associated with depression, forgetfulness, speech problems (dysarthria), anxiety and emotional lability as a result of interruption of prefrontal-subcortical circuits by infarcts.[7] Some patients with VaD later develop a short gait and urge incontinence, depending on the areas in the brain affected. Major depression is more common and severe in patients with VaD than in those with AD.
The course of vascular dementia is generally shorter than that of AD. Average survival from diagnosis is four to five years and patients often die of a cardiovascular or cerebrovascular event. Management will, therefore, involve preventing such events and patients are typically prescribed an anticoagulant, an antihypertensive and a lipid regulator. Prevention will also include lifestyle changes, such as stopping smoking and eating a healthier diet. Antipsychotics (eg, risperidone or olanzapine) and antidepressants are also used. Some clinicians prescribe drugs licensed to treat AD to patients with VaD.
Box 2: Homocysteine levels
Homocysteine (Hcy) is an amino acid derived from methionine catabolism. Hcy plasma levels rise with age. High plasma Hcy levels have been associated with poor cognition and dementia.[5] It has been related to neurodegenerative disease in a number of human studies with evidence indicating that cell redox potential is altered, leading to the protein accumulation and apoptosis.
Hcy and its primary metabolite have been shown to act as agonists at N-methyl Daspartate (NMDA) receptors which, in turn, are implicated in neurotoxicity via various proposed biochemical changes. Hcy metabolism depends on the presence of B vitamins (B12 and B6 and folate [B9]) and it is unclear whether hyperhomocysteinaemia causes neurotoxicity or B vitamin deficiency is the primary cause of neurodegeneration.
The neurotoxic effects of hyperhomocysteinaemia can be ameliorated by folate, NMDA receptor antagonists (eg, memantine) and various antioxidants (see Box 3).
Box 3: Vitamin E
The concept that dementia development is related to oxidative stress has led to interest in the use of antioxidant compounds, such as vitamins E and C, to prevent or treat it. The brain appears to be particularly affected by oxidative damage because of its high oxygen and energy consumption, the relatively poor activity of antioxidant enzymes and the fact that cells are composed of a high proportion of polyunsaturated fatty acids that are susceptible to free radical damage.[6]
Vitamin E has been shown to reduce oxidative stress and potentially alter cellular protein metabolism, helping to maintain the integrity of the central nervous system. Some studies have shown that development of cognitive decline correlates with low serum vitamin E levels and that supplementation with 2,000iu of vitamin E in patients with mild to moderate dementia slows the progression of the disease significantly in terms of the primary outcomes of death, a need for residential care and loss of independence with activities of daily living. However, this evidence is countered by studies suggesting that disease progression is not altered.
At present it would be controversial to state that supplementation will slow or reverse disease progression.
Dementia with Lewy bodies
Dementia with Lewy bodies (DLB) accounts for 15 to 20 per cent of dementia cases. Disagreement exists as to exactly what DLB is — the disease seems to lie somewhere between AD and Parkinson’s disease. DLB is differentiated clinically from AD by looking for a specific pattern of symptoms (see Box 4). A (probable) diagnosis can only be made if patients have two core features or one core feature with supportive features. These features should be present without a history of stroke or other cause of cognitive decline.
DLB has distinct microscopic features: patients present with Lewy bodies and amyloid plaques in the subcortical and cortical regions of the brain. Neurofibrillary tangles are less commonly seen. Lewy bodies are eosinophilic cytoplasmic inclusions that are distinct from neurofibrillary tangles. They contain a protein called alpha-synuclein, which is normally responsible for synaptic plasticity. Alphasynuclein also appears to regulate the size of presynaptic vesicular pools of neurotransmitters and, therefore, influences neurotransmission. In DLB, heavily phosphorylated alpha-synuclein becomes cross linked to form the insoluble complexes that are Lewy bodies. Lewy bodies also contain a variety of inflammatory markers (eg, interleukins), proteases,βΑ and lipids. Lewy body development is accompanied by neuronal loss with specific deficits in cholinergic and dopaminergic neurotransmission.
There is no drug to slow down or prevent DLB and management involves medicines to improve quality of life. For example, hallucinations and depression (symptoms that can be particularly troublesome in DLB) can be treated with atypical antipyschotics and selective serotonin reuptake inhibitors, respectively.
Some patients are prescribed medicines that are licensed for AD (eg, rivastigmine) or Parkinson’s disease (eg, levodopa), or enrolled in clinical trials.
Box 4: Examples of features of DLB
Core features:
- Progressive cognitive decline affective social and occupational functioning
- Recurrent detailed hallucinations
- Spontaneous parkinsonism (eg, dystonia)
Supportive features:
- Falls
- Loss of consciousness (eg, fainting)
- Sleep disorders (specifically, altered phases of rapid eye movement sleep)
- Depression
Frontotemporal dementias
Frontotemporal dementias (FTD; eg, Pick’s disease) represent 5 to 20 per cent of dementias. Onset is most commonly between 45 and 65 years of age and patients have a mean survival of seven to nine years from diagnosis. FTD occurs with equal frequency in males and females.[8]
In the initial stages of the disease, both intellect and memory are relatively well preserved so FTD is commonly misdiagnosed. The frontotemporal lobe influences mood and behaviour so patients generally present with changes in personality, including loss of inhibitions, inserting inappropriate objects into the mouth (hyperorality), obsessional disorders and apathy.
Language disturbances, including reduction in verbal fluency and loss of meaning of words, and motor symptoms can also occur and FTDs are largely divided into two groups; those with predominant behavioural disturbances and those with language deterioration. Familial FTD has been linked to a genetic mutation on chromosome 17 resulting in the formation of abnormal tau proteins in a similar manner to AD.
The progression of frontotemporal dementia cannot be slowed. Drugs licensed for AD, such as galantamine or rivastigmine, are sometimes prescribed.
Huntington’s disease
Huntington’s disease (HD) is an autosomal dominant disorder with an average course of 15 to 20 years. It typically affects patients between 30 and 50 years of age and has a prevalence of between four and seven per 100,000 worldwide.[9] The main and initial symptom of HD is “fidgety” movements (chorea) but cognitive decline, attention deficit and depression follow. Development of bipolar disease and schizophrenia is common in patients with HD.
The gene mutation responsible has been identified as a polymorphic trinucleotide repeat in chromosome 4p. This is responsible for producing mutant forms of the Huntington protein. This is normally found in cell cytoplasm but, in HD, it is seen in nuclei. The mechanism of neurodegeneration as a result of this genetic mutation is unclear and is currently the subject of research.The net result is a deficit in gamma-aminobutyric acid (GABA; the main inhibitory neurotransmitter in the central nervous system) and increased dopaminergic activity.
HD is pharmacologically managed with tetrabenazine (for motor symptoms) and antidepressants or antipsychotics where needed.
HIV dementia
HIV dementia (HIVD) is likely to become more common, considering the rise in HIV infection. It is estimated that up to 30 per cent[10] of patients with HIV will develop HIVD. HIVD is a subcortical dementia where patients present with poor cognitive flexibility, reduced response times, apathy and emotional lability. The pathological mechanism involved in HIVD appears to be apoptosis in neuronal cells as a result of various neurotoxic chemicals (proteases, cytokines etc) shed from infected macrophages and glial cells within the CNS. There is some evidence to suggest that treatment with antiretroviral agents could reduce the incidence of HIVD and, in patients who have developed the dementia, possibly stop or slow progression.
Conclusion
Numerous pathological processes affecting various areas of the brain result in the development of dementia. An understanding of these mechanisms and of the symptoms caused by neurodegeneration in different brain areas is important in understanding how we treat these conditions in practice now, as well as in the future.
Patients with dementia are likely to experience compliance issues and this is clearly an area in which pharmacists can play a role. However, it is also important for pharmacists to understand what having dementia means for the lives of patients and carers so that they can give support when needed, be it signposting or explaining medication.
Dementia is already relatively common but is bound to increase with our rapidly ageing population.
Andrew Husband, MSc, MRPharmS, and Alan Worsley, PhD, MRPharmS, are senior lecturers at Sunderland University
References
1. World Health Organization. International classification of diseases. 10th revision. Geneva: World Health Organization;1992.
2. Thomas H, Rai G. The aetiology and pathology of dementia. Hospital Pharmacist 2001;8:33–40.
3. Ferri CP, Prince M, Crayne C, Brodaty H, Fratiglioni L, Ganguli M et al. Global prevalence of dementia:a Delphi consensus study. Lancet 2005;366:2112–7.
4. Loeb C, Meyer JS. Vascular dementia: still a debatable entity? Journal of the Neurological Sciences 1996;143:31–40.
5. Seshadri S, Beiser A, Selhub J, Jacques P, Rosenberg IH, D’agostino R et al. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. New England Journal of Medicine 2002;346:476–83.
6. Cherubini A, Martin A, Andres-Lacueva C, Di Iorio A, Lamponi M, Mecocci P et al. Vitamin E levels, cognitive impairment and dementia in older persons: the InCHIANTI study. Neurobiology of Ageing 2005;26:987–94.
7. Roman GC, Erkinjuntti T, Wallin A, Pantoni L, Chui H. Subcortical ischaemic vascular dementia. Lancet Neurology 2002;1:426–36.
8. Sjoegren M, Andersen C. Frontotemporal dementia — a brief review. Mechanisms of Ageing and Development 2006;127:180–7.
9. Gelder M, Mayou R, Cowen P (editors). Shorter Oxford textbook of psychiatry. 4th ed. Oxford: Oxford University Press; 2001.
10. McArthur JC, Hoover DR, Bacellar H, Miller EN, Cohen BA, Becker JT et al. Dementia in AIDS patients: incidence and risk factors. Multicentre AIDS cohort Study. Neurology 1993;43:2245–56.