Alzheimer’s disease is a progressive disorder. After symptoms first appear, life expectancy is around 8-10 years. Memory is typically the first problem, then difficulties with cognition and planning become marked, speech and language are affected, and patients may experience altered personality and behaviour. Eventually, individuals are unable to care for themselves (see PDF of graphic).
Two main processes are thought to drive degeneration: intracellular accumulation of a protein called tau and extracellular deposition of a protein called beta-amyloid (Aβ), leading to formation of neurofibrillatory tangles and amyloid plaques. Although most people develop some tangles and plaques as they age, those with symptoms of Alzheimer’s disease tend to develop far more.
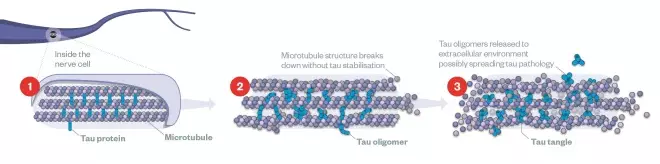
Tau and tangles
(1) Inside the healthy neuron, parallel microtubules provide the cell’s transport system for nutrition and key materials to be delivered. Tau protein act as scaffolding for the microtubules, keeping them in place.
(2) Hyperphosphorylation of tau leads to formation of tau oligomers. These oligomers clump together with other molecules and form tangles.
(3) By distorting the spacing of microtubules, tangles impair axonal transport thus affecting the nutrition of axon terminals and dendrites, eventually leading to cell death.
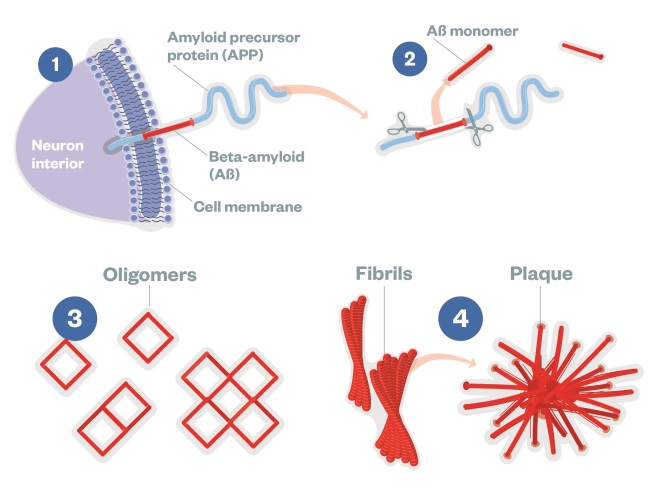
Beta-Amyloid
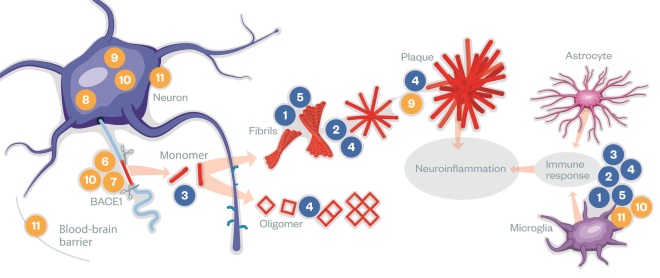
(1) APP is a transmembrane protein, made by neurons and other brain cells. (2) Aβ is cut from APP by the enzymes β-secretase (also called BACE1) and γ-secretase releasing Aβ monomers into the extracellular environment. (3) Once clipped, Aβ monomers can assemble in at least two ways: one pathway leads to soluble oligomers of Aβ 42, which are small enough to enter synapses. (4) Another pathway for Aβ monomers is to assemble into insoluble fibrils and larger plaques, which can provoke harmful inflammation.
Biological targets
Licensed drugs treat symptoms by regulating levels of the neurotransmitters acetylcholine and glutamate, which are important for neuronal communication. In contrast, drugs in development aim to inhibit the mechanisms that lead to the build-up of plaques and tangles characteristic of the condition by targeting Aβ, tau or the inflammation caused by plaques and tangles.
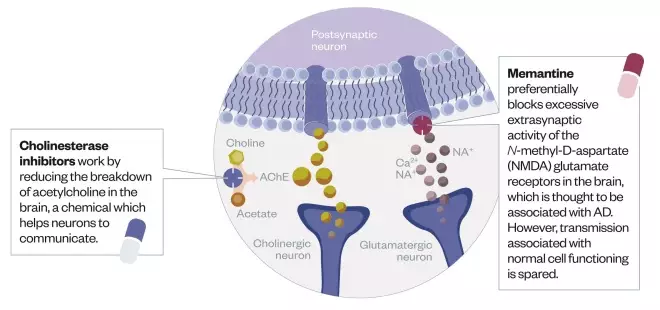
Licensed treatments
Three cholinesterase inhibitors are currently licensed to treat Alzheimer’s disease symptoms, as well as one N-methyl-D- aspartate (NMDA) blocker.
Pipeline drugs
Monoclonal antibodies (blue) aim to clear toxic Aβ either directly or through microglia or astrocyte activation. However, antibodies have limited bioavailability in the brain and can induce immunogenicity. Small molecules (yellow) aim to reduce Aβ production, prevent tau aggregation or target neuroinflammation. They have better bioavailability in the brain than the antibodies. See table below for breakdown of drugs
| Monoclonal antibodies | |
|---|---|
| 1 | Chugai Pharmaceutical/Hoffmann-La Roche’s gantenerumab is a fully human IgG1 monoclonal antibody that recognises primarily fibrils of Aβ. It stimulates microglia to clear amyloid plaques. |
| 2 | Biogen’s aducanumab is a high affinity, fully human IgG1 monoclonal antibody that binds aggregated forms of Aβ, not monomers. It stimulates microglia to clear amyloid plaques. |
| 3 | Eli Lilly’s solanezumab is a humanised monoclonal IgG1 antibody, which recognises soluble monomers of Aβ, not fibrils. It stimulates microglia to clear toxic amyloid monomers. |
| 4 | Genentech’s crenezumab is a fully human IgG4 monoclonal antibody, which recognises multiple forms of aggregated Aβ, including oligomers and fibrils and amyloid plaques with high affinity, and monomers with low affinity. It stimulates microglia enough to clear Aβ, but not enough to induce an inflammatory response. |
| 5 | Biogen/Eisai’s BAN2401 is a humanised IgG1 monoclonal antibody that selectively binds to large, soluble Aβ protofibrils and stimulates their clearance by microglial cells. |
| Small molecules | |
|---|---|
| 6 | Gamma-secretase modulators , such as Merck’s GSM-1 and Eisai’s E2012, reduce the cleavage of APP that produces the most toxic peptide, Aβ 42, while still allowing the enzyme to cleave its other substrates. |
| 7 | Merck’s MK-8931 and AstraZeneca’s AZD3293 both inhibit BACE-1, so interfere with production of Aβ. |
| 8 | TauRx Therapeutics’s LTMX is a tau aggregation inhibitor that reduces levels of aggregated or misfolded tau proteins. |
| 9 | Two licensed drugs (names not released) have been found to inhibit the enzyme PERK, which is thought to switch off production of all new proteins in the brain in response to accumulation of amyloid plaques. New proteins are necessary for neuronal plasticity and memory consolidation. |
| 10 | T3D Therapeutics’s T3D-959 is a dual nuclear receptor agonist, which has the potential to inhibit both the Aβ and tau pathways. |
| 11 | Pfizer/TransTech’s azeliragon is an antagonist of the cell surface receptor RAGE, which can lead to inflammation and oxidative damage when occupied. RAGE also mediates transport of Aβ from the blood into the brain. |
Drug development
Alzheimer’s disease (AD) was first identified by German doctor Alois Alzheimer in 1906 but it was not until 1993 that the first drug to target the symptoms of Alzheimer’s was approved by the US Food and Drug Administration (FDA). Despite a 99.6% attrition rate for AD drugs between 2002 and 2012, there is hope for current pipeline drugs being tested in prodromal and mild AD, with the aim of stopping disease development rather than treating symptoms.
1993: Warner-Lambert’s tacrine (Cognex) receives FDA approval for mild-to-moderate AD but is later discontinued.
1996: Eisai’s donepezil (Aricept) receives FDA approval for mild-to-moderate AD. This was extended to include severe AD in 2006.
2000: Novartis’s rivastigmine (Exelon) receives FDA approval for mild-to-moderate AD.
2000: Shire’s galantamine (Reminyl) receives European Medicines Agency (EMA) approval for mild-to-moderate AD.
2002: Forest Laboratories’s memantine (Ebixa) receives EMA approval for moderate-to-severe AD.
2012: Two phase III trials of Eli Lilly’s monoclonal antibody solanezumab in patients with mild-to-moderate AD do not reach their endpoints.
2012: Phase II trial of Biogen/Eisai’s monoclonal antibody BAN2401 begins recruiting 800 people with early stage AD. Estimated completion date is July 2018.
2012: Two phase III trials of TauRx Therapeutics’s LMTX , a second-generation tau aggregation inhibitor, begin in patients with mild and mild-to-moderate AD. Study results are expected in 2016.
2013: Phase II trial of Genentech’s monoclonal antibody crenezumab starts in patients with preclinical AD, with results due in 2020. Two additional phase 2 trials in mild-to-moderate AD are ongoing.
2013: Phase III trial begins to assess safety and efficacy of MSD’s BACE inhibitor MK-8931 compared with placebo in patients with prodromal AD. The trial is set to run until 2018.
2014: Actavis/Adamas’s donepezil/memantine combination product (Namzaric) receives FDA approval for moderate-to-severe AD.
2015: Biogen reports a dose-dependent reduction in amyloid plaque and slowing of cognitive decline for monoclonal antibody aducanumab in an interim analysis of phase 1b data for prodromal or mild AD patients.
2015: New “delayed-start” analysis of negative phase III solanezumab trials suggests it may slow progression of mild AD.
- This graphic was amended on 17 August 2015 to show that the RAGE receptor mediates transport of beta-amyloid across the blood-brain barrier.